
Abstract
The complete mitochondrial genome (mitogenome) of Bactrocera caudata (Diptera: Tephritidae: Dacinae) was sequenced and annotated. The mitochondrial genome is 15,311 bp (GenBank No. MT180970), containing 72.7% A + T (A 39.1%, C 17.0%, G 10.3%, and T 33.6%), which is the classical structure for insect mitogenome. All PCGs were started with ATN (ATA/ATG/ATT/ATC) and terminated with TAA except ND3, which ends with TAG. Additionally, the phylogenetic tree confirmed that Bactrocera caudata was closely related to Bactrocera dorsalis and Bactrocera correcta. The current study would enrich the mitogenomes of the fruit flies.
Bactrocera caudata has a Paleartic and Oriental distribution. It occurs in India, Sri Lanka, Myanmar, Thailand, Vietnam, China, Malaysia, Brunei and Indonesia. It is a pest of commercial and edible flowers. Specimens of B. caudate had been reared from flowers of pumpkin Cucurbita moschata in Peninsular Malaysia (Phaik-Eem et al. Citation2012). The identification of B. caudata is mainly based on morphology and partial sequence. Presently, we have sequenced and determined the complete mitochondrial genome (mitogenome) for the first time, which might facilitate future studies on evolution of the subfamily Dacinae.
The genome DNA was extracted from male adult of B. caudata which was collected in Jinzhu town vineyard, Guizhou province, China (E106°38′26″, N26°30′26″), in August 2018, and the voucher specimen’s genome DNA is deposited in the Research Center for Guizhou Characteristic Fruits and its Products of Mountainous Regions, Guizhou Light Industry Technical College, label number is GCZX-108. These sequences were assembled using Geneious Primer (Kearse et al. Citation2012), version 10.2.3 (http://www.geneious.com/). Additionally, all tRNAs were found by MITOS server (http://mitos.bioinf.uni-leipzig.de/index.py) (Bernt et al. Citation2013) and tRNA scan-SE server (Lowe and Chan Citation2016) for annotation. The neighbor-joining (NJ) tree was constructed to investigate the molecular taxonomic position of B. caudata based on nucleotide sequences of 13 protein-coding genes and 2 rRNA genes using MEGA 6.0 (Tamura et al. Citation2013) from alignments created by the MAFFT (Katoh and Standley Citation2013).
The complete mitogenome of B. caudata is 15,311 bp (GenBank No. MT180970), containing 22 transfer RNA genes (tRNAs, 1,482 bp), 13protein-coding genes (PCGs, 11,295 bp), 2 ribosomal RNA genes (rRNAs, 2,086 bp), and 1 non-coding region (Control region, 388 bp). The whole genome contained 39.1% A, 17.0% C, 10.3% G, and 33.6%T, showing an obvious A + T bias (72.7%). The AT-skew (0.0757) for the whole mitogenome is slightly positive, but negative for GC-skew (–0.2454). All PCGs were started with ATN (ATA/ATG/ATT/ATC) and terminated with TAA excepting ND3, which ends with TAG
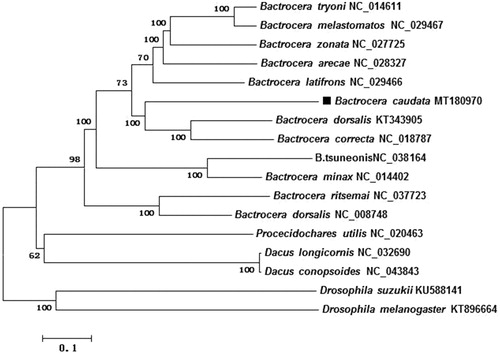
The phylogenetic relationships of B. caudata were reconstructed using the neighbor-joining (NJ) method with 1000 bootstrap replicates based on concatenated nucleotides of the 13 PCGs and 2 rRNAs with 13,381 bp, Dosophila suzukii and Drosophila melanogaster were used as outgroup (). The phylogenetic tree confirms that B. caudata was closely related to Bactrocera dorsalis and Bactrocera correcta. Presently, the studies recording B. caudata were limited, and we believe that our data could be useful for further study.
Figure 1. Neighbor-joining (NJ) phylogenetic tree of Bactrocera proprediaphora based on concatenated nucleotides of the 13 PCGs and 2 rRNAs by MEGA 6.0.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Bernt M, Donath A, Jühling F, Externbrink F, Florentz C, Fritzsch G, Pütz J, Middendorf M, Stadler PF. 2013. MITOS: improved de novo metazoan mitochondrial genome annotation. Mol Phylogenet Evol. 69(2):313–319.
- Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 30(4):772–780.
- Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, et al. 2012. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 28(12):1647–1649.
- Lowe TM, Chan PP. 2016. Trna scan-SE On-line: integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res. 44(W1):W54–W57.
- Phaik-Eem L, Ji T, Wayan S, Praphathip E, Hio SY. 2012. Distinct genetic lineages of Bactrocera caudate (Insecta:Tephritidae) revealed by CO1 and 16S DNA sequences. PLoS One. 7(5):e37276.
- Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. 2013. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol. 30(12):2725–2729.