
Abstract
Clematis fruticosa Turcz. is an erect shrubby plant with important ecological and economical value, which usually occurs in dry or desert areas. The complete chloroplast genome of C. fruticosa was assembled from Illumina pair-end sequence reads. The whole plastome is 159,684 bp in length and presents a quadripartite structure consisting of two copies of inverted repeat (IR) regions (31,045), a large single copy (LSC) region (79,461 bp) and a small single copy (SSC) region (18,133 bp). The plastome of C. fruticosa encodes a total of 137 genes, including 92 protein-coding genes, 1 pseudogene, 36 tRNA genes and 8 rRNA genes. The overall GC content of C. fruticosa plastome is 38.0%. A maximum likelihood (ML) phylogenetic analysis revealed that C. fruticosa was collectively sister to a clade of Clematis tangutica and Clematis aethusifolia with high support.
Clematis fruticosa is an erect shrubby plant belonging to Clematis Sect. Fruticella, which usually occurs in dry or desert areas of northern China (Wen-Tasi and Liang-Qian Citation2004). Considering the controversial taxonomical delimitation, discontinuous geographical distribution and xerophilous feature (He et al. Citation2018), Clematis Sect. Fruticella is an interesting group for phylogeny and drought resistance study. Here, we report and characterize the complete chloroplast (cp) genome of C. fruticosa (GenBank accession number: MT083932), which will provide genomic data for the related studies.
An individual of C. fruticosa was sampled from Jiufeng Mountain (Baotou, Inner Mongolia, China). The specimen was deposited at the herbarium of Baotou Teachers’ College (Accession number: JFS-2019-09-B02GMTXL). Total genomic DNA was isolated from silica-dried leaf material using modified CTAB method (Doyle Citation1987), and then was sequenced using an Illumina Hiseq 2500 platform at Biomarker Technologies Inc. (Beijing, China). Firstly, all of the raw reads were trimmed using NGS QC Toolkit_v.2.3.3 with the default parameters set (Patel and Jain Citation2012). Reference-guided assembly was then used to reconstruct the cp genomes with the programs MIRA 4.0.2 (Chevreux et al. Citation2004) and MITObim v1.7 (Hahn et al. Citation2013). In the process, cp genomes of Clematis terniflora (NC_028000) and Clematis macropetala (NC_041477) were used as reference genomes. The complete cp genome was annotated using the program DOGMA (Wyman et al. Citation2004), and then manually corrected by comparing them with the complete cp genomes of some published Clematis species in GENEIOUS R10 (Biomatters Ltd., Auckland, New Zealand).
The complete cp genome of C. fruticosa is 159,684 bp in length with a typical quadripartite structure. The large single copy (LSC) region, small single copy (SSC) region and inverted repeat (IRa and IRb) regions are 79,461 bp, 18,133 bp and 31,045 bp, respectively. The assembled plastome encodes a total of 137 genes, consisting of 92 protein-coding genes, 1 pseudogene, 36 tRNA genes, and 8 rRNA genes. The overall GC content of C. fruticosa cp genome is 38.0%, and the corresponding values in LSC, SSC and IR regions are 36.3%, 31.4%, and 42.0%, respectively.
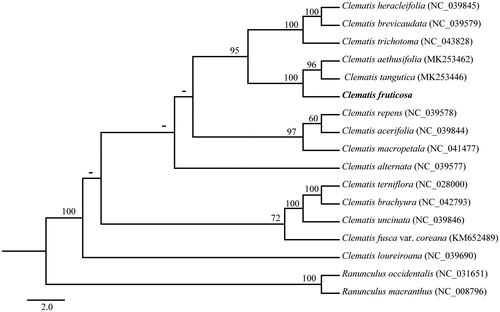
The phylogenetic tree was constructed based on 15 complete cp genomes of Clematis species and two Ranunculus species as outgroup. All of the 17 complete cp genome sequences were aligned using MAFFT (Katoh and Standley Citation2013) with default parameter and maximum likelihood (ML) analysis was performed using RAxML v8 (Stamatakis Citation2014) with 1000 bootstrap replicates. The phylogenetic tree indicated that C. fruticosa was collectively sister to a clade of Clematis tangutica and Clematis aethusifolia with high support ().
Figure 1. The phylogenetic tree based on 17 complete chloroplast genome sequences. The number on each node indicates the bootstrap value. Dash denotes the bootstrap value lower than 50%.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability
The data that support the findings of this study are openly available in NCBI at https://www.ncbi.nlm.nih.gov/, reference number (MT083932).
Additional information
Funding
References
- Chevreux B, Pfisterer T, Drescher B, Driesel AJ, Müller WE, Wetter T, Suhai S. 2004. Using the miraEST assembler for reliable and automated mRNA transcript assembly and SNP detection in sequenced ESTs. Genome Res. 14(6):1147–1159.
- Doyle JJ. 1987. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem Bull. 19:11–15.
- Hahn C, Bachmann L, Chevreux B. 2013. Reconstructing mitochondrial genomes directly from genomic next-generation sequencing reads – a baiting and iterative mapping approach. Nucleic Acids Research. 41(13):e129–e129.
- He J, Liu HJ, Xie L. 2018. Research advances of Clematis Sect. Fruticella (Ranunculaceae). J Nanjing Forestry University (Nat Sci Ed.). 42(1):156–162.
- Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 30(4):772–780.
- Patel RK, Jain M. 2012. NGS QC Toolkit: a toolkit for quality control of next generation sequencing data. PLoS One. 7(2):e30619.
- Stamatakis A. 2014. RAxML Version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 30(9):1312–1313.
- Wen-Tasi W, Liang-Qian LI. 2004. A revision of Clematis sect. Fruticella (Ranunculaceae). J Syst Evolut. 43(3):193–209.
- Wyman SK, Jansen RK, Boore JL. 2004. Automatic annotation of organellar genomes with DOGMA. Bioinformatics. 20(17):3252–3255.