
Abstract
The complete chloroplast genome of Corydalis conspersa was sequenced and assembled. It is a circular form of 187,810 bp in length, which was separated into four distinct regions, a large single-copy (LSC) of 92,280 bp, a small single-copy region (SSC) of 780 bp, two inverted repeats (IR) of 47,375 bp. After annotation, a total of 144 genes were predicted, of which, 98 encode proteins, 8 rRNA, 38 tRNA. The evolutionary history, inferred using neighbour-joining method, indicates C. conspersa was grouped within Papaveraceae, and comprised a clade with Lamprocapnos spectabilis with 100% BS value.
Corydalis conspersa Maxim, belonging to Papaveraceae, is a alpine herb plant, possessing yellow tepals with brown spots. It is distributed mainly in southwest Gansu, southwest Qinghai, northwest and west Sichuan, east and middle Tibet in China, at an elevation from 3500–5300 m (Que et al. Citation2017). For containing various alkaloids, plants of Corydalis are generally used as fehrifuge and detoxicant in traditional Chinese medicine (Zeng et al. Citation1987; Dae-Keun et al. Citation2000). Corydalis conspersa, like other species in Corydalis, is used extensively in traditional Tibet medicine in curing of ache, inflammation, anxiety, etc. (Fang et al. Citation1984; Wang et al. Citation2019). In previous studies, although chemical composition of this plant have been well documented, the knowledge of the genetics or molecular biology about this important medical plant is still less known. In this study, we report the complete chloroplast genome of C. conspersa.
A sample from Qilian mountains (36°24′3″N, 101°22′6″E) in Qinghai province was used for sequencing. Voucher specimen (HNWP-336861) was deposited in the Herbarium, Northwest Institute of Plateau Biology (HNWP). Total genomic DNA was extracted from 100 mg fresh leaves using a modified CTAB method (Murray and Thompson Citation1980). Paired-end Libraries with an average length of 350 bp were constructed. Sequencing was performed on the Illumina Novaseq platform (Shenzhen Huitong biotechnology Co. Ltd., Shenzhen, China). The complete cp genome was assembled with the de novo assembler SPAdes (Bankevich et al. Citation2012). Gene annotation was performed via plan. (Huang and Cronk Citation2015).
The complete cp genome of C. conspersa (GenBank accession no. MN843953) has a typical quadripartite form of 187,810 bp in length and composed of a large single-copy region (LSC, 92,280 bp), a small single-copy region (SSC, 780 bp), two inverted repeats (IR, 47,375 bp). GC content of the genome is 37.9%. A total of 144 genes were predicted on this cp genome, of which, 98 encode proteins, 8 rRNA, 38 tRNA.
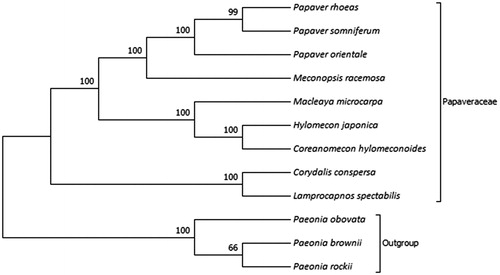
Phylogenetic analysis was performed based on complete cp genomes of C. conspersa and other eight related species reported in Papaveraceae, three species in Ranunculaceae as out-groups. The sequences were aligned using MAFFT (Katoh et al. Citation2002) and trimAl was employed to remove ambiguously aligned sites (Capella-Gutierrez et al. Citation2009). The evolutionary history was inferred using neighbour-joining method in MEGA7.0 (Kumar et al. Citation2016). Bootstrap (BS) values were calculated from 1000 replicate analysis (). As expected, C. conspersa was grouped within Papaveraceae and comprised a clade with Lamprocapnos spectabilis with 100% BS value. The complete cp genome of C. conspersa will be helpful for further studies on population genetics, taxonomy or resources protection.
Figure 1. NJ phylogenetic tree based on 12 species chloroplast genomes was constructed using MEGA7.0. Numbers on each node are bootstrap from 1000 replicate. Accession numbers: Papaver rhoeas NC_037831.1; Papaver somniferum NC_029434.1; Papaver orientale NC_037832.1; Meconopsis racemosa NC_039625.1; Macleaya microcarpa NC_039623.1; Hylomecon japonica MK251463.1; Coreanomecon hylomeconoides KT274030.1; Corydalis conspersa MN843953; Lamprocapnos spectabilis NC_039756.1; Paeonia obovata NC_026076.1; Paeonia brownii NC_037880.1; Paeonia rockii NC_037772.1.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability
The data that support the findings of this study are available from the corresponding author, [J.W], upon reasonable request.
Additional information
Funding
References
- Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, et al. 2012. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 19(5):455–477.
- Capella-Gutierrez S, Silla-Martinez JM, Gabaldon T. 2009. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics. 25(15):1972–1973.
- Dae-Keun K, Jae-Soon E, Tae-Yong S, Dong-Ok E, Jong-Pil L. 2000. Benzo[c] phenanthridine alkeloids from Corydalis incisa. Arch Pharm Res. 23(6):589–591.
- Fang QC, Mao L, Weng QM. 1984. Chemical study of alkaloids from Corydalis conspersa. Planta Med. 50(1):25–27.
- Huang DI, Cronk Q. 2015. Plann: a command-line application for annotating plastome sequences. Appl Plant Sci. 3(8):1–3.
- Katoh K, Misawa K, Kuma K, Miyata T. 2002. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 30(14):3059–3066.
- Kumar S, Stecher G, Tamura K. 2016. MEGA7: Molecular Evolutionary Genetics Analysis version 7.0 for bigger datasets. Mol Biol Evol. 33(7):1870–1874.
- Murray MG, Thompson WF. 1980. Rapid isolation of high molecular weight plant DNA. Nucl Acids Res. 8(19):4321–4326.
- Que S, Da LJ, Zeng QY, Lin PC, Chen WD. 2017. Chemical constituents of the volatile component from Corydalis conspersa Maxim by supercritical CO2 fluid extraction and their antibacterial activity. Chin J NewDrugs. 26(2):203–207.
- Wang Y, Wang M, Wang Z, Lin PC. 2019. Determination of the content of putropin in Corydalis Conspersa Mazim by HPLC. J Shanxi Datong Univ. 35(2):1–3.
- Zeng WG, Liang WZ, Tu G. 1987. Chemical study of alkaloids from Corydalis bungeana. Planta Med. 53(5):418–420.