
Abstract
Anogeissus acuminata (Roxburgh ex Candolle) Guillemin et al. is an Endangered and dominant species of deciduous forests distributed in the Mekong valley of southwest China and adjacent Indo-China Peninsula. Here we assembled and annotated the complete chloroplast (cp) genome. It is 159,993 bp in length and encodes 84 protein-coding genes, 37 transfer RNA (tRNA) genes and eight ribosomal RNA (rRNA) genes. This chloroplast genome sequencing offers genetic background for conservation and phylogenetic studies.
Anogeissus is a small genus distributed in tropical Africa and Asia, only one species occurs in China A. acuminata is edemic to tropical Asia, as a dominant tree deciduous forests distributed in the Mekong valley of southwest China and adjacent Indo-China Peninsula under 700 m in altitudes (Chen and Turland Citation2007).
The species was listed as Endangered in China by Shun (Fu Citation1992). In the present study, the completed chloroplast genome sequence of A. acuminata is reported contributing for better understanding its evolution and population genetics, and also providing significant information for the phylogeny of Combretaceae.
Genomic DNA was extracted from fresh leaves of a seedling of A. acuminata from Mengkuang, Nuozhadu, Lancang, Yunnan, China (100°31′1.14″E, 22°29′58.50″N, 651 m, in the valley; Xu Tongmei et al.19090, 2019-1-29; KUN), the total genomic DNA was isolated according to a modified CTAB method (Doyle and Doyle Citation1987). Those leaves were stored in the refrigerator at -80 °C. Total genome DNA of A. acuminata was sequenced by Illumina Hiseq 2500 Sequencing System (Illumina, Hayward, CA, USA) to construct the shotgun library and assembled through the NOVOPlasty software (Dierckxsens et al. Citation2017).The low quality sequences were filtered out Using CLC Genomics Workbench v8.0 (CLC Bio, Aarhus, Denmark) and then reconstructed the chloroplast genome by using MITObim v1.8 (University of Oslo, Oslo, Norway; Kaiseraugst, Switzerland) (Hahn et al. Citation2013). The complete chloroplast genome of A. acuminata was annotated in Geneious ver. 10.1 (http://www.genei ous.com, Kearse et al. Citation2012) and then submitted to GenBank (accession no. MT242598). The genome annotation was performed by aligning with the cp genomes of relatively related species.
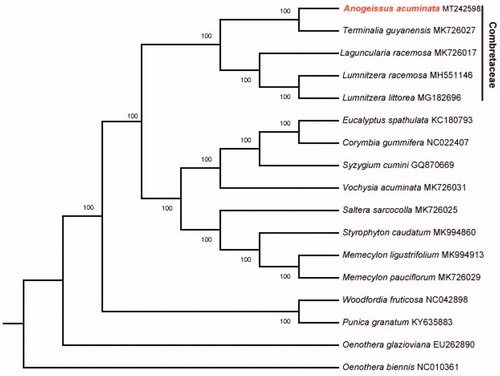
The size of chloroplast genome of A. acuminata is 159,993 bp, including a large single-copy (LSC) region of 88,430 bp and a small single-copy (SSC) region of 18,871 bp separated by a pair identical inverted repeat regions (IRs) of 26,346 bp each. A total of 129 genes were successfully annotated containing 84 protein-coding genes, 37 tRNA genes and 8 rRNA genes. GC content of the whole genome, IRs, LSC and SSC regions are 37.0, 43.0, 34.7 and 30.7%, respectively. GC content of IRs region is the highest. Twenty genes contain one intron, while 3 genes have two introns. The complete chloroplast genome sequence of A. acuminata and other species from Combretaceae were used to construct phylogenetic tree (). The sequences were initially aligned using MAFFT (Katoh and Standley Citation2013) and then visualized and manually adjusted using BioEdit (Hall Citation1999). Take the plastome of Oenothera biennis (GenBank: NC_010361) as an out-group, a maximum likelihood analysis was performed with RAxML version 8 program (Alexandros Citation2014) using 1000 bootstrap. The result shows the position of A. acuminata from Myrtales, which is consistent with previous molecular results (Conti et al.Citation1997; Bayly et al. Citation2013; Gu et al. Citation2019).
Figure 1. Maximum-likelihood phylogenetic tree for A. acuminata based on 16 complete chloroplast genomes. The number on each node indicates bootstrap support value.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability
The data that newly obtained at this study are available in the NCBI under accession number of MT242598.
Additional information
Funding
References
- Alexandros S. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 30(9):1312–1313.
- Bayly MJ, Rigault P, Spokevicius A, Ladiges PY, Ades PK, Anderson C, Bossinger G, Merchant A, Udovicic F, Woodrow IE, et al. 2013. Chloroplast genome analysis of Australian eucalypts – Eucalyptus, Corymbia, Angophora, Allosyncarpia and Stockwellia (Myrtaceae). Mol Phylogenet Evol. 69(3):704–716.
- Chen J, Turland NJ. 2007. Combretaceae. In: Wu ZY, Raven PH, editors. Flora of China. Vol. 13. Beijing: Science Press and Missouri Botanical Garden Press; p. 314.
- Conti E, Litt A, Wilson PG, Graham SA, Briggs BG, Johnson LAS, Sytsma KJ. 1997. Interfamilial relationships in Myrtales: molecular phylogeny and patterns of morphological evolution. Syst Bot. 22(4):629–647.
- Dierckxsens N, Mardulyn P, Smits G. 2017. NOVOPlasty: de novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 45(4):e18.
- Doyle JJ, Doyle JL. 1987. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem Bull. 19:11–15.
- Fu LK. 1992. China plant red data book-rare and Endangered plants. Beijing: Science Press.
- Gu C, Ma L, Wu Z, Chen K, Wang Y. 2019. Comparative analyses of chloroplast genomes from 22 Lythraceae species: inferences for phylogenetic relationships and genome evolution within Myrtales. BMC Plant Biol. 19(1):281.
- Hahn C, Bachmann L, Chevreux B. 2013. Reconstructing mitochondrial genomes directly from genomic next-generation sequencing reads—a baiting and iterative mapping approach. Nucleic Acids Res. 41(13):e129–e129.
- Hall TA. 1999. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp Ser. 41:95–98.
- Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 30(4):772–780.
- Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, et al. 2012. Geneious *Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 28(12):1647–1649.