
Abstract
Populus trinervis, which is a unique plant for China. The complete chloroplast genome sequence of Populus trinervis was characterized from Illumina pair-end sequencing. The chloroplast genome of P. trinervis was 156,415 bp in length, containing a large single-copy region (LSC) of 84,805 bp, a small single-copy region (SSC) of 16,505 bp, and two inverted repeat (IR) regions of 27,554 bp. The overall GC content is 36.70%, while the corresponding values of the LSC, SSC, and IR regions are 34.5, 30.5, and 42.0%, respectively. The genome contains 131 complete genes, including 86 protein-coding genes (62 protein-coding gene species), 37 tRNA genes (29 tRNA species), and 8 rRNA genes (4 rRNA species). The Neighbour-joining phylogenetic analysis showed that P. trinervis and P. hopeiensis clustered together as sisters to other Populus species.
Introduction
Populus is a genus of deciduous flowering plants, which was traditionally divided into six sections based on leaf and flower characters. Populus trinervis is grouped into Populus section Tacamahaca, and its leaves are 4–7 cm long, 2.5–5 cm wide. Being native to China, P. trinervis is found in Sichuan provinces. Given to both natural and artificial inter-specific hybrids, the classification of polar is very difficult. The genetic relationships and evolutionary history of poplar are still poorly investigated (Zheng et al. Citation2017; Hou et al. Citation2018). Populus trinervis plays an important ecological role in boreal and temperate forests, serving as wildlife habitats and watersheds; they can dominate riparian forests, but are ecologically adaptable. Populus trinervis has high intraspecific polymorphism, adaptability to different environments, combined with a relatively small genome size. Consequently, P. trinervis represents an excellent model for understanding how different evolutionary forces have sculpted the variation patterns in the genome during the process of population differentiation and ecological speciation (Neale and Antoine Citation2011). Moreover, we can develop conservation strategies easily when we understand the genetic information of P. trinervis. In the present research, we constructed the whole chloroplast genome of P. trinervis and understood many genome variation information about the species, which will provide beneficial help for population genetics studies of P. trinervis.
The fresh leaves of P. trinervis were collected from Sichuan (30°05′N, 102°54′E). Fresh leaves were silica-dried and taken to the laboratory until DNA extraction. The voucher specimen (SMQY001) was laid in the Herbarium of Chongqing University of Arts and Sciences and the extracted DNA was stored in the −80 °C refrigerator of the Key Laboratory of College of Landscape Architecture and Life Science. We extracted total genomic DNA from 25 mg silica-gel-dried leaf using a modified CTAB method (Doyle Citation1987). The whole-genome sequencing was then conducted by Biodata Biotechnologies Inc. (Hefei, China) with Illumina Hiseq platform. The Illumina HiSeq 2000 platform (Illumina, San Diego, CA, USA) was used to perform the genome sequence. We used the software MITObim 1.8 (Hahn et al. Citation2013) and metaSPAdes (Nurket al. Citation2017) to assemble chloroplast genomes. We used P. tremula (GenBank: NC_027425) as a reference genome. We annotated the chloroplast genome with the software DOGMA (Wyman et al. Citation2004), and then corrected the results using Geneious 8.0.2 (Campos et al. Citation2016) and Sequin 15.50 (http://www.ncbi.nlm.nih.gov/Sequin/).
The complete chloroplast genome of P. trinervis (GenBank accession number MT482538) was characterized from Illumina pair-end sequencing. T The complete chloroplast genome sequence of P. trinervis was characterized from Illumina pair-end sequencing. The chloroplast genome of P. trinervis was 156,415 bp in length, containing a large single-copy region (LSC) of 84,805 bp, a small single-copy region (SSC) of 16,505 bp, and two inverted repeat (IR) regions of 27,554 bp. The overall GC content is 36.70%, while the corresponding values of the LSC, SSC, and IR regions are 34.5, 30.5, and 42.0%, respectively. The genome contains 131 complete genes, including 86 protein-coding genes (62 protein-coding gene species), 37 tRNA genes (29 tRNA species), and 8 rRNA genes (4 rRNA species).
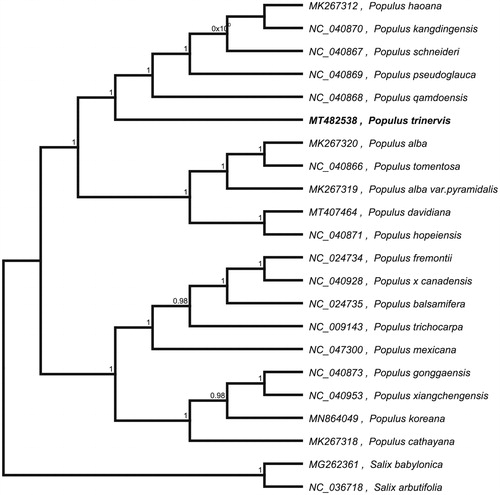
To confirm the phylogenetic location of P. trinervis within the family of Populus, we used the complete chloroplast genomes sequence of P. trinervis and 19 other related species of Populus and S. babylonica and Salix arbutifolia as outgroup to construct phylogenetic tree. The 14 chloroplast genome sequences were aligned with MAFFT (Katoh and Standley Citation2013), and then the Neighbour-joining tree was constructed by MEGA 7.0 (Kumar et al. Citation2016). The results confirmed that P. trinervis was clustered with P. davidiana ().
Figure 1. Neighbour-joining (NJ) analysis of P. trinervis and other related species based on the complete chloroplast genome sequence.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability
The data that support the findings of this study are openly available in National Center for Biotechnology Information (NCBI) at https://www.ncbi.nlm.nih.gov, accession number MT482538.
Additional information
Funding
References
- Campos FS, Kluge M, Franco AC, Giongo A, Valdez FP, Saddi TM, Brito WMED, Roehe PM. 2016. Complete genome sequence of porcine parvovirus 2 recovered from swine sera. Genome Announc. 4(1):e0162701615.
- Doyle J. 1987. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem Bull. 19(1):11–15.
- Hahn C, Bachmann L, Chevreux B. 2013. Reconstructing mitochondrial genomes directly from genomic next-generation sequencing reads—a baiting and iterative mapping approach. Nucleic Acids Res. 41(13):e129–e129.
- Hou Z, Wang Z, Ye Z, Du S, Liu S, Zhang J. 2018. Phylogeographic analyses of a widely distributed Populus trinervis: further evidence for the existence of glacial refugia of cool‐temperate deciduous trees in northern. Ecol Evol. 8(24):13014–13026.
- Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 30(4):772–780.
- Kumar S, Stecher G, Tamura K. 2016. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol. 33(7):1870–1874.
- Neale DB, Antoine K. 2011. Forest tree genomics: growing resources and applications. Nat Rev Genet. 12(2):111–122.
- Nurk S, Meleshko D, Korobeynikov A, Pevzner PA. 2017. metaSPAdes: a new versatile metagenomic assembler. Genome Res. 27(5):824–834.
- Wyman SK, Jansen RK, Boore JL. 2004. Automatic annotation of organellar genomes with DOGMA. Bioinformatics. 20(17):3252–3255.
- Zheng H, Fan L, Milne RI, Lei Z, Wang Y, Mao K. 2017. Species delimitation and lineage separation history of a species complex of aspens in China. Front Plant Sci. 8:375.