
Abstract
Malus toringoides belongs to the subfamily Amygdaloideae of Rosaceae, which is an endemic species in China. It has significant ornamental, economic, and ecological value. Herein, we assembled the complete chloroplast genome of Malus toringoides using the next-generation sequencing technology. The total length of the complete chloroplast genome was 160,093 base pair (bp), consisting of one large single-copy (LSC) region with a sequence length of 88,177 bp, one small single-copy (SSC) region which sequence length is 19,194 bp, and a pair of inverted repeat regions (IRs, 26361 bp). Besides, the complete chloroplast genome contained 128 genes, namely 83 protein-coding genes (PCGs), 37 tRNA genes (tRNA), and 8 rRNAgenes (rRNA), the GC content was 36.6%. The phylogenetic relationship among species in genus Malus is closely related, especially the phylogenetic relationship among Malus angustifolia, Malus prattii, Malus micromalus, Malus prunifolia, Malus baccata, Malus hupehensis and Malus toringoides. Furthermore, phylogenetic relationship of Malus toringoides in Malus was closely related to Malus hupehensis. Our study affords important genetic information for further researches on the related species.
Introduction
Malus, a genus, belongs to the Rosaceae family, which includes approximately 35 species all over the world. The majority of the species grow in the temperate regions of the Northern Hemisphere. There are 23 species in China and most of them possess important ornamental, economic, and ecological value (Tang et al. Citation2015). Among them, Malus toringoides is one of the seven endemic species in China (Qian Citation2005; Wu and Hong Citation2009). Apples are suitable for hillside terraces, plain wilderness and loess hills, with an elevation of 50–2500 meters (Wu and Hong Citation2009). Due to these unique merits in physiological features, the species in this genus are ideal garden greening and important honey source for insects. Contributing to the widespread hybridization between species within genus Malus, the species relationship among this genus is confused. Therefore, we hope to explore the phylogenetic relationship in Malus based on the sequences of chloroplast genome.
Total genomic DNA was extracted from the silica gel dried and clean leaves of Malus toringoides sampled from Wuhan Botanical Garden (China; N30°31′21.62″, E114°25′34.99″). Meanwhile, the voucher specimen (POC529082) was deposited in the herbarium, Institute of Botany, Chinese Academy of Sciences (PE). Total DNA was extracted via modified cetyltrimethyl ammonium bromide (mCTAB) method (Li et al. Citation2013). An NGS library was constructed and sequenced at Beijing Novogene Bioinformatics Technology Co., Ltd (Beijing) on Illumina HiSeq2500 platform using pair-end 150 bp (PE150) strategy. Twenty millions sequence reads were obtained and chloroplast genome reads were sorted out and the genome was assembled de novo using SPAdes 3.9 (Bankevich et al. Citation2012). Finally, the assembled chloroplast genome was then annotated on Plann with Malus angustifolia (No.NC045410) as reference and corrected with Sequin (Huang and Cronk Citation2015). And then, the annotated chloroplast genome sequences were submitted to GenBank (accession number: MT483999).
The complete chloroplast genome of Malus toringoides is 160,093 bp in length. It includes two copies of inverted repeats (IRs, 26361 bp), a large single-copy (LSC, 88177 bp), and a small single-copy (SSC, 19194 bp) regions. A total of 128 genes were annotated, containing 83 protein-coding genes (PCGs), 37 transfer RNA genes (tRNA), and 8 ribosomal RNA genes (rRNA). Overall GC content was 36.6% and those in LSC, SSC, and IR regions were 34.2%, 30.4%, and 42.7%, respectively.
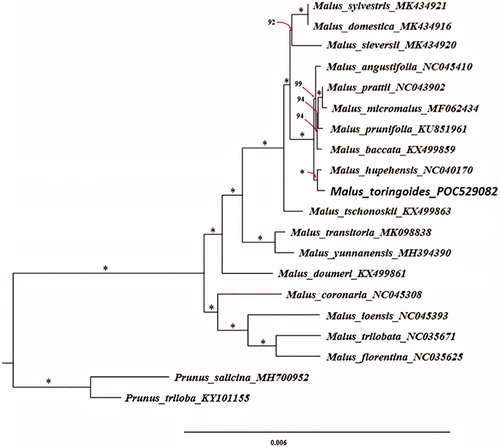
A phylogenetic analysis was carried out using whole chloroplast genome sequences of Malus toringoides and other 17 chloroplast genome sequences of species in Malus which could be downloaded from the NCBI database. Meanwhile, two species from the Prunus were used as outgroups. Multiple sequences alignment was executed by MAFFT (Katoh and Standley Citation2013). ModelFinder was used for model selection according to the Bayesian information criterion (BIC) (Kalyaanamoorthy et al. Citation2017). A maximum likelihood (ML) tree with 1000 bootstrap replicates were inferred by IQ-TREE (Nguyen et al. Citation2015). The phylogenetic result indicates that the phylogenetic relationship among species in genus Malus is closely related, especially among species Malus angustifolia, Malus prattii, Malus micromalus, Malus prunifolia, Malus baccata, Malus hupehensis and Malus toringoides. The relationship within genus Malus can be well distinguished using whole chloroplast genome data. Malus toringoides is most closely related to Malus hupehensis (100% ultrafast bootstrap support, ) (Hoang et al. Citation2018). This result would be beneficial to potential studies on phylogenetics of the genus and related group in Rosaceae.
Figure 1. Phylogenetic tree based on plastid genomes using the ML method. Ultrafast bootstrap (UFBoot) values are shown above the nodes, with 1000 bootstrap replicates. *Represents that this result is 100% supported.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
The data that support the findings of this study are openly available in NCBI GenBank database at (https://www.ncbi.nlm.nih.gov) with the accession number is MT483999, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Additional information
Funding
References
- Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, Pyshkin AV, et al. 2012. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 19(5):455–477.
- Hoang DT, Chernomor O, von Haeseler A, Minh BQ, Vinh LS. 2018. UFBoot2: improving the ultrafast bootstrap approximation. Mol Biol Evol. 35(2):518–522.
- Huang DI, Cronk Q. 2015. Plann: a command-line application for annotating plastome sequences. Appl Plant Sci. 3(8):1500026.
- Kalyaanamoorthy S, Minh BQ, Wong TKF, von Haeseler A, Jermiin LS. 2017. ModelFinder: fast model selection for accurate phylogenetic estimates. Nat Methods. 14(6):587–589.
- Katoh K, Standley D. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 30(4):772–780.
- Li J, Wang S, Yu J, Wang L, Zhou S. 2013. A modified CTAB protocol for plant DNA extraction. Chin Bull Bot. 48:72–78.
- Nguyen LT, Schmidt HA, von Haeseler A, Minh BQ. 2015. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. 32(1):268–274.
- Qian G. 2005. Taxonomic Study on Malus Mill. [Ph.D. thesis]. Nanjing, China: Nanjing Forestry University.
- Tang F, Ding Z, Ren J, Shi D, Liu Z, Jin X, Guo R. 2015. Species and varieties of ornamental crabapple in China. J Anhui Agri Sci. 43(16):190–195.
- Wu Z, Hong D. 2009. Rosaceae. Flora of China. Vol. 36. Beijing: Sciences Press.