
Abstract
In this study, we first obtained the complete mitochondrial genome of Neilupotamon xinganense (Decapoda: Brachyura: Potamoidea). The genome is 16,965 bp in length and typically consists of 37 genes (13 protein-coding genes, 22 tRNAs genes, two rRNAs genes, and one putative control region). In addition, the mitogenome has 20 non-coding regions ranging from 1 to 683 bp in length. This study provides DNA data for further researches on population genetics and phylogenetics.
The genus Neilupotamon is endemic to mainland China, including N. papilionaceum, N. physalisum, N. sinense, and N. xinganense. The complete mitochondrial genome of N. sinense had been reported (Zhang et al. Citation2020). In this study, we report for the first time the complete mitochondrial genome of N. xinganense.
An adult specimen of N. xinganense was collected from Dayuan Village, Zhongfeng Town, Ziyuan County, Guilin City, Guangxi Province, China in 2018 (N25.9224°, E110.6572°). The sample has been deposited in the Laboratory Specimen Library of Freshwater Crustacean Decapoda and Paragonimus, School of Basic Medical Sciences, Nanchang University, Nanchang, Jiangxi, China, and National Parasite Germplasm Resources Specimen Library of China with a catalog number of NCUMCP4068. The sample was stored in 95% ethanol prior to extraction at room temperature before sequence analyses. Genomic DNA extraction, sequencing, gene annotation, and phylogenetic analyses were performed according to the method described by Plazzi et al. (Citation2013). The Bayesian Inference (BI) method was performed using MrBayes vers. 3.2 (Ronquist et al. Citation2012), with best model GTR + I + G selected by jModelTest vers.2.1.7. The maximum-likelihood (ML) method was performed using MEGA 6 (Tamura et al. Citation2013).
The complete genome of N. xinganense is 16,965 bp in length (GenBank accession number: MN117718) and contains 13 protein-coding genes (PCGs), 22 tRNA genes, two rRNA genes, and one control region (CR) as the typical metazoan mitochondrial genome. However, in N. xinganense, we can only clearly annotate the location of 21 kinds of tRNA, the location of tRNAIle is still in doubt. We try to search for traces of missing tRNA by inferring the evolutionary history of gene structure rearrangement. By comparing with the gene sequence of relative species, we can basically determine that the location of tRNAIle should be located in the 86 bp intergenic region between 12S rRNA gene and tRNAMet, and roughly determine that 64 bp is a special tRNAIle, and its anticodon is also transformed from the normal GAT to TAT.
Among the 37 genes, 23 genes are encoded by H-strand and 14 genes are encoded by L-strand. The total length of coding gene is 14,738 bp, and the total length of non-coding region is 2227 bp. There are 20 non-coding regions with a length of 1–683 bp, the longest non-coding region is between tRNAGln and COX2. There are seven overlapped regions with a total length of 21 bp and a length of 1–7 bp, among which the longest overlapped region is between ND4 and ND4L. Among 13 protein-coding genes (PCGs), ND1, ND4, ND4L, and ND5 are encoded by L-strand, and the other nine are encoded by H-strand. The start codon of COX1, COX2, COX3, ATP8, ND2, ND3, ND4L, ND5, and CYTB is ATG, the start codon of ATP6, ND1, and ND6 is ATA, and the start codon of ND4 is GTG. For stop codon, except ND5 is TAT and COX2 is incomplete T––, all the other genes are TAA. The average A + T content of PCGs is 64.93%. ND4L has the highest A + T content of 68.65%, CYTB has the lowest A + T content of 60.16%.
The length of 22 tRNA genes is between 62 bp (tRNACys) and 72 bp (tRNAVal). Fourteen tRNA genes are encoded by H-strand and the rest by L-strand. All tRNA genes except for tRNASer(AGN) and tRNAIle showed typical cloverleaf structure. The 16S rRNA gene and 12S rRNA gene are encoded by L-strand, with length of 1330 bp and 826 bp respectively. The 16S rRNA gene is located between tRNA Leu(TAG) and tRNAVal, whereas the 12S rRNA gene is located between CYTB and tRNAIle. The longest non-coding region, the control region, lies between COX2 and tRNAGln, with a length of 683 bp and the A + T content of 72.18%.
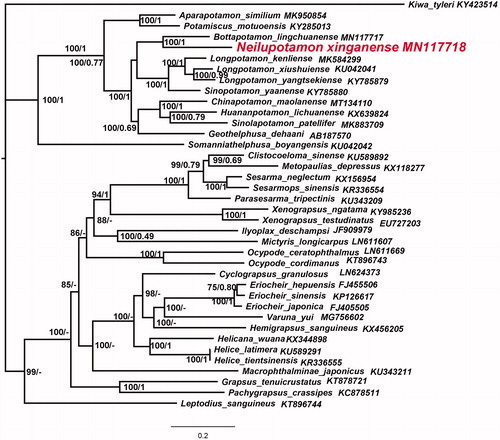
The phylogenetic position of N. xinganense in mitogenome relative to other Brachyuran mitogenomes is determined by applying the BI and ML methods on 13 PCGs (). Phylogenetic analysis showed that the two species Bottapotamon lingchuanense and N. xinganense, which have unique gene sequence, gather into one branch, and form sister branches with the evolutionary branch composed of four species of Sinopotamon and Longpotamon.
Figure 1. Phylogenetic maximum-likelihood (ML) tree of Neilupotamon xinganense and related brachyurans based on 13 PCGs nucleotide sequences from the mitochondrial genome. Kiwa tyleri serves as the outgroup. The numbers at the internodes are Bayesian inference (BI) bootstrap proportions and ML posterior proportions. The differences between the ML and BI trees are indicated by ‘–’. The scale bars represent genetic distance.
Acknowledgements
The authors thank Song-bo Wang for giving useful advice on writing.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
The data that support the findings of this study are openly available in GenBank of NCBI at https://www.ncbi.nlm.nih.gov, reference number MN117718.
Additional information
Funding
References
- Plazzi F, Ribani A, Passamonti M. 2013. The complete mitochondrial genome of Solemya velum (Mollusca: Bivalvia) and its relationships with Conchifera. BMC Genomics. 14(1):409.
- Ronquist F, Teslenko M, van der Mark P, Ayres DL, Darling A, Hohna S, Larget B, Liu L, Suchard MA, Huelsenbeck JP. 2012. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol. 61(3):539–542.
- Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. 2013. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol. 30(12):2725–2729.
- Zhang Z, Xing YH, Cheng JJ, Pan D, Lv L, Cumberlidge N, Sun HY. 2020. Phylogenetic implications of mitogenome rearrangements in East Asian potamiscine freshwater crabs (Brachyura: Potamidae). Mol Phylogenet Evol. 143:106669.