
Abstract
Hirundo is the most species-rich genus of the passerine swallow family (Hirundinidae) and has a cosmopolitan distribution. Here we report the complete, annotated mitochondrial genomes for 25 individuals from 10 of the 14 extant Hirundo species; these include representatives from four subspecies of the barn swallow, H. rustica. Mitogenomes were conserved in size, ranging from 18,500 to 18,700 base pairs. They all contained 13 protein-coding regions, 22 tRNAs, a control region, and large and small ribosomal subunits. Phylogenetic analysis resolved most of the relationships between the studied species and subspecies which were largely consistent with previously published trees. Several new relationships were observed within the phylogeny that could have only been discovered with the increased amount of genetic material. This study represents the largest Hirundo mitochondrial phylogeny to date, and could serve as a vital tool for other studies focusing on the evolution of the Hirundo genus.
Introduction
The genus Hirundo comprises 14 species of swallows and has a cosmopolitan distribution spanning every continent except Antarctica. Of these species, the barn swallow (H. rustica) is the most widespread, breeding throughout Eurasia and North America, and overwintering across the Southern Hemisphere (Brown and Brown Citation1999). Eleven of the Hirundo species reside primarily in Africa, and three others inhabit the Pacific. Members of Hirundo build mud-cup nests in a variety of habitats (e.g. cliffs, anthills, human dwellings, bridges) and are highly adapted to aerial feeding. Among members of the genus Hirundo, variation in melanin-based plumage color is thought to be a prominent aspect of phenotypic differences. In addition to differences in plumage color among members of the genus Hirundo, length of outer tail streamers and body size tend to be helpful aspects of phenotype for identifying species (Dor et al. Citation2010). Differences in these character traits are often minimal, which presents challenges to taxonomic efforts (Sheldon et al. Citation2005; Dor et al. Citation2010).
Dor et al. published a species tree of all members of the genus Hirundo using one nuclear gene and six mitochondrial DNA genes with the purpose of understanding lineage diversification and phylogeographic relationships within this clade (Dor et al. Citation2010). This was significant as this was the only phylogeny of the Hirundo genus using more than 1000 base pairs of genetic material at the time. An earlier paper by (Sheldon et al. Citation2005) presented the entire swallow family, Hirundinidae, and used even fewer genes, thus a lower base pairs count, for its phylogeny. The ability to sequence and assemble entire mitogenomes is now more feasible than in the past due to reduced financial costs and improved computational accessibility; re-evaluating the phylogeny using more genetic data is now a more achievable task.
In the present study, we revisited the evolutionary relationships among members of the genus Hirundo by assembling complete mitochondrial genomes of 10 species. Genomes were assembled de novo from Illumina shotgun sequence data. These mitochondrial genomes offer greater resolution and support for some aspects of the previous Dor et al. (Citation2010) Hirundo mtDNA gene tree and also offer insight to a new topology.
Materials and methods
Sample collections
Samples were collected from both museum breast tissues and field collection blood samples as depicted in .
Table 1. Complete collection of samples used in analysis.
DNA extraction and sequencing
Genomic DNA was extracted using a QIAamp DNA Blood Mini kit, following instructed blood or tissue protocol. Genomic libraries were then prepared using Nextera® XT DNA library prep kits (Illumina®), and each sample was barcoded using unique dual index adapters Nextera® i5 and i7 by the University of Colorado’s BioFrontiers Institute Next-Generation Sequencing Facility in Boulder Colorado. Samples that passed quality control were processed for paired end 150 base pair reads on the Illumina Novaseq S4 sequencer (800 raw data/lane) at Novogene®.
Mitochondrial genome assembly
The whole genome data were aligned to the Hirundo rustica gutturalis reference mitogenome (GenBank accession KP148840.1) with Samtools function bwa-mem using default settings (Li Citation2013). Only the reads that aligned to the mitochondria were retained for use in downstream assembly (this subsetting of the data improved assembly performance). Reads were trimmed of adapters and low-quality reads using Trimmomatic v0.39 (Bolger et al., Citation2014)) with the following parameters: Illuminaclip: NexteraPE-PE.fa:2:20:10 Leading:20 Trailing:20 Sliding window:4:15 Minlen:100. De novo assembly of trimmed reads into scaffolds was performed with SPAdes v3.11.1 (Bankevich et al., Citation2012). The relative position, order, and orientation of scaffolds were determined by comparison with available H. rustica rustica and H. rustica erythrogaster reference genomes available on GenBank (GenBank accessions KP148840.1, KX398931.1). When multiple contigs represented the same genomic region, contig selection was based on maintaining approximate consistency in read coverage across contigs. Properly ordered contigs were then combined by trimming overlapping sequences. Gaps between scaffolds were filled by tiling from raw or trimmed reads.
Mapping and error correction
Assembled genomes were then aligned to reference Hirundo mitochondrial genomes (GenBank accessions KP148840.1, KX398931.1) with Zpicture, which allowed visualization of structural differences between the reference and our mitochondrial genomes (Ovcharenko et al., Citation2004). Assembled genomes were also compared according to relatedness for structural agreement following the work of Dor et al. (Citation2010). Samtools tView was then used to identify possible SNPs (single nucleotide polymorphisms) or Indels (insertions and/or deletions) in the genome; modifications were made if mapped reads supported these assembly errors/variants (Li Citation2011). Completed draft assemblies were compared using Multiple Alignment using Fast Fourier Transform (MAFFT) ver. 7 (Katoh and Standley Citation2013). Large discrepancies in assemblies based on alignment to neighboring or same species were taken into account when making changes to the draft genome sequences.
Annotation
Genomic features were annotated using MITOS and MITOS2 (Bernt et al., Citation2013). Accuracy of these annotations was checked by comparison to annotations from Mitofish (Iwasaki et al., Citation2013) and tRNAscan-SE 2.0 (Lowe and Eddy Citation1997). Annotations were further verified using BLAST to compare the nucleotide and translated protein sequences of our mitogenomes against the same reference mitogenomes as used in the alignment step (GenBank accessions KP148840.1, KX398931.1). Sequence and annotation information was entered in NCBI’s Sequin 15.50 software for final submission to GenBank. In some cases, Sequin identified errors in assembly (e.g. premature stop codons), which were then fixed via the methods described above.
Phylogenetic analysis
A maximum likelihood phylogenetic hypothesis was established using sequence data from the annotated whole mitochondrial genomes. The taxa list consists of 26 individuals from 10 species: Delichon urbicum (n = 4), Hirundo atrocaerulea (n = 1), Hirundo dimidiata (n = 2), Hirundo tahitica (n = 2), Hirundo neoxena (n = 2), Hirundo albigularis (n = 1), Hirundo nigrita (n = 2), Hirundo smithii (n = 3), Hirundo angolensis (n = 1), Hirundo aethiopica (n = 2), Hirundo rustica tytleri (n = 1), Hirundo rustica rustica (n = 1), Hirundo rustica transitiva (n = 2), and Hirundo rustica savignii (n = 1). Sequences were aligned in MAFFT (v7) in a CLUSTAL format (Katoh and Standley Citation2013). RAxML (v.8.2.10) was used to infer the phylogenetic hypothesis (Stamatakis Citation2014). To analyze node support 1000 Bootstrap replicates were used in a GTR GAMMA maximum likelihood search. The tree was rooted to Delichon urbicum, per the previous Hirundinidae mtDNA phylogeny, which shows D. urbicum being the closest relative apart from Ptyonoprogne fuligula (Sheldon et al., 2005).
Results and discussion
Genome annotation
Mitochondrial genome content across birds has been studied intensively, and has been found to generally include the following features, which were present in all of our assemblies: 13 protein-coding genes (ND1, ND2, ND3, ND4, ND4l, ND5, ND6, COI, COII, COIII, ATPase6, ATPase8, and Cytb), 22 tRNAs, and the large and small rRNA of the ribosome (Boore, Citation1999). We found incomplete stop codons for COXIII and NAD4, which is a common feature of avian mitogenomes (Anmarkrud and Lifjeld Citation2017). Post-transcriptional polyadenylation of RNA is a likely completion mechanism of these stop codons, however, studies of transcriptomic data will be necessary to verify this hypothesis. The mitochondrial genomes also exhibit characteristic compactness, with a maximum separation of 40 base pairs between genes. The vast majority of genes were encoded on the + strand. Of the 37 identified mitochondrial genes, only nine were encoded on the − strand, grouped into two operons and two standalone genes. Additionally, the genes were ordered identically across all sampled species, including the outgroup D. urbicum. The length of the highly variable rrnS and rrnL genes were relatively consistent across species, with rrnS measuring ∼1000 base pairs across species, and rrnL measuring ∼1600 base pairs.
Phylogenetic comparison
In recreating the Hirundo genus phylogeny, several differences can be observed between the previous Dor et al. phylogeny and the current complete mitochondrial genome phylogeny topologies.
We found two main differences between our Hirundo phylogeny and those preceding it (Sheldon et al., 2005; Dor et al., Citation2010); both differences are within the ‘Barn Swallow’ clade (figure 1). First, we found an unresolved polytomy within the ‘Barn Swallow clade associated with the H. albigularis, H. smithii, and H. nigrita taxons: the hypothesis of H. nigrita, H. angolensis, H. aethiopica, H. r. tytleri, H. r. rustica, H. r. transitiva, H. r. savignii being nested within H. smithii, as previously suggested, was not well supported (bootstrap support = 49/100)(figure1). This indicates uncertainty in the position of H. albigularis relative to H. nigrita and H. smithii. Geographically, H. albigularis and H. smithii species boundaries overlap in the southern regions of Africa, making a moderate amount of gene flow a potential causal factor for the polytomy, apart from the lack of time since divergence (Maddison and Knowles Citation2006) (figure 1). Second, while H. angolensis was originally nested within the H. rustica clade, our data show the opposite pattern of H. rustica along with H. aethiopia placed within the H. angolensis clade ().
Figure 1. Maximum-likelihood phylogenetic hypothesis using GTR-GAMMA model parameter. Taxa (n = 26) rooted to Delicum urbicum. Outgroup: Delicum urbicum. Blue Clade: Hirundo atrocarulea. PB Clade (Pearl-Breasted Clade): Hirundo dimidiata. Pacific Clade: Hirundo tahitica & Hirundo neoxena. Barn Swallow Clade: Hirundo albigularis, Hirundo smithii, Hirundo nigrita, Hirundo angolensis, Hirundo aethiopia, Hirundo rustica tytleri, Hirundo rustica rustica, Hirundo rustica transitiva, and Hirundo rustica savignii.
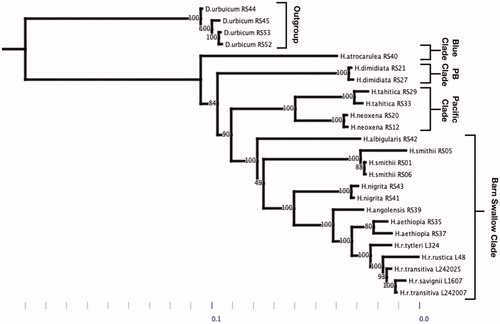
Our findings also clarify a previously unresolved node connecting the ‘Blue Swallow’ and ‘Pearl-Breasted Swallow’ clades with the joint ‘Pacific Swallow’ and ‘Barn Swallow Clades’ (Dor et al., Citation2010). We found that the latter two clades are sister to the ‘Pearl-Breasted Swallow’ clade (bootstrap support = 84/100). The previous phylogeny (Dor et al., Citation2010) had suggested this relationship; however, a low bootstrap value of 54/100 prevented a definitive conclusion.
In addition to an updated tree topology, our whole mitogenome data also provide greater support for branch length values. Higher support within branch length is generally associated with more genetic data that agrees with the topology patterns and high levels of congruency (Wiens et al., Citation2008).
It is interesting that the whole mitogenomes presented here have clarified certain aspects of the Hirundo phylogeny while at the same time introducing an unresolved polytomy. An exact topology remains elusive even at the level of whole mitochondrial genomes. We suspect this is attributable to how closely relate these species are. Despite the benefits mitogenomes offer for phylogenetic studies, it is important to remember mitochondrial genes are linked and ultimately represent a single, matrilineal locus. We anticipate additional nuclear genomic markers will be of use in resolving all nodes of the Hirundo phylogeny. Future phylogenomic studies of Hirundo will investigate whether the nuclear gene tree agrees with the mtDNA tree presented here. The potential ILS associated with the polytomy between H. albigularis, H. smithii, and the H. nigrita clade will also be addressed using a phylonetwork approach during the nuclear genome analysis.
Acknowledgments
Thanks to the students of the University of Colorado, Boulder, Genomics class of 2019 for their hard work. Special thanks to the collection managers Dr. John Bates (Field Museum), Dr. Kristof Zyskowski (Yale Peabody Museum), Dr. Mariel Campbell (University of New Mexico museum), and Dr. Donna Dittmann (Louisiana State University Museum) for donating specimens from their museums for this and other experiments. For field collected specimens, we thank Dr. Elizabeth S.C. Scordato, Dr. Amanda Hund and Dr. Joanna Hubbard.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
The authors confirm that the data supporting the findings of this study are available within the article [accession information for GenBank in ]. No supplementary material was needed.
Additional information
Funding
References
- Anmarkrud JA, Lifjeld JT. 2017. Complete mitochondrial genomes of eleven extinct or possibly extinct bird species. Mol Ecol Resour. 17(2):334–341.
- Bankevich A, Nurk S, Antipov D, Gurevich A A, Dvorkin M, Kulikov A S, Lesin V M, Nikolenko S I, Pham S, Prjibelski A D, et al. 2012. SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing. J. Comput. Biol. 19(5):455–477. doi:10.1089/cmb.2012.0021.
- Bernt M, Donath A, Jühling F, Externbrink F, Florentz C, Fritzsch G, Pütz J, Middendorf M, Stadler P F. 2013. MITOS: Improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenet. Evol. 69(2):313–319. doi:10.1016/j.ympev.2012.08.023.
- Bolger A M, Lohse M, Usadel B. 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 30(15):2114–2120. doi:10.1093/bioinformatics/btu170.
- Boore J L. 1999. Animal mitochondrial genomes. Nucleic Acids Res. 27(8):1767–1780. doi:10.1093/nar/27.8.1767.
- Brown CR, Brown MB. 1999. Barn swallow (Hirundo rustica). Birds North Am. 452:32.
- Dor R, Safran RJ, Sheldon FH, Winkler DW, Lovette IJ. 2010. Phylogeny of the genus Hirundo and the Barn Swallow subspecies complex. Mol Phylogenet Evol. 56(1):409–418.
- Held P, Buehrer L. 2003. The Synergy™ HT: a unique multi-detection microplate reader for HTS and drug discovery. J Assoc Lab Autom. 8(2):44–49.
- Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 30(4):772–780.
- Iwasaki W, Fukunaga T, Isagozawa R, Yamada K, Maeda Y, Satoh TP, Sado T, Mabuchi K, Takeshima H, Miya M, et al. 2013. MitoFish and MitoAnnotator: a mitochondrial genome database of fish with an accurate and automatic annotation pipeline. Mol Biol Evol. 30(11):2531–2540.
- Li H. 2011. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics. 27(21):2987–2993.
- Li, Heng. “Aligning sequence reads, clone sequence and assembly contigs with BWA-MEM.” arXiv preprint arXiv:1303.3997 (2013).
- Lowe TM, Eddy SR. 1997. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25(5):955–964.
- Maddison WP, Knowles LL. 2006. Inferring phylogeny despite incomplete lineage sorting. Syst Biol. 55(1):21–30.
- Ovcharenko I, Loots GG, Hardison RC, Miller W, Stubbs L. 2004. zPicture: dynamic alignment and visualization tool for analyzing conservation profiles. Genome Res. 14(3):472–477.
- Sheldon F H, Whittingham L A, Moyle R G, Slikas B, Winkler D W. 2005. Phylogeny of swallows (Aves: Hirundinidae) estimated from nuclear and mitochondrial DNA sequences. Mol. Phylogenet. Evol. 35(1):254–270. doi:10.1016/j.ympev.2004.11.008.
- Sheldon FH, Winkler DW. 1993. Intergeneric phylogenetic relationships of swallows estimated by DNA-DNA hybridization. Auk. 110(4):798–824.
- Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 30(9):1312–1313.
- Wiens JJ, Kuczynski CA, Smith SA, Mulcahy DG, Sites JW, Townsend TM, Reeder TW. 2008. Branch lengths, support, and congruence: testing the phylogenomic approach with 20 nuclear loci in snakes. Syst Biol. 57(3):420–431.