
Abstract
Trachycarpus fortunei (Hook.) H. Wendl. (Fam.: Palmae; Gen.: Trachycarpus) is an evergreen tree that is widely distributed in China. In this study, T. fortunei complete chloroplast (cp) genome was assembled. The total cp genome size of T. fortunei was 158,613 bp in length, containing a large single-copy region of 86,422 bp, a small single-copy region of 17,847 bp, and a pair of inverted repeat regions of 27,172 bp. The overall GC-content of T. fortunei cp genome was 37.21%. It encodes a total of 109 unique genes, including 79 protein-coding genes, 26 tRNA genes, four rRNA genes. Twelve genes contain a single intron and 11 genes have two introns. Phylogenetic analysis results reveal that T. fortunei was closely related to Chamaerops humilis.
Trachycarpus fortunei (Hook.) H. Wendl. (Fam.: Palmae; Gen.: Trachycarpus) is an evergreen tree that is widely distributed in China, where its leaf sheath fiber is often used as a rope and its unopened flower buds, also known as ‘brown fish,’ are edible and consumed (Yunfa Citation2005).
The cp genome contains a large amount of genetic information and has highly conservative characteristics. Trachycarpus fortunei seeds were collected in Guiding County, Guizhou Province, China (E: 107°07′42″, N: 26°13′19″). The seeds were germinated and nursed in the laboratory (the seed specimen is accessible at the Institute for Forest Resources & Environment of Guizhou, Guizhou University (accessions No. TF-001-2)), total genome DNA of collected annual new needles was extracted with EasyPure® Plant Genomic DNA Kit (TransGen Biotech, Beijing, China). Total DNA was used to generate libraries with an average insert size of 400 bp, which were sequenced using the Illumina NovaSeq platform. Trachycarpus fortunei cp genome sequence was assembled by SPAdes (Bankevich et al. Citation2012) and A5-miseq (Coil et al. Citation2015) fragment assembly. The complete cp genome of T. fortunei was annotated in CPGAVAS2 (http://47.96.249.172:16019/analyzer/home, Shi et al. Citation2019). The annotated cp genome sequence has been deposited into the Genbank (accession number: MT712077).
Trachycarpus fortunei cp genome exhibited a general quadripartite structure of plants, with two reverse repeated regions (IRa and IRb) of 27,172 bp in length. The repeat regions divided the genome into two single-copy regions, SSC and LSC with17,847 and 86,422 bp, respectively. The overall GC-content of the T. fortunei cp genome is 37.21%. It encodes a total of 109 unique genes, including 79 protein-coding genes, 26 tRNA genes, four rRNA genes. Twelve genes (rps16, atpF, rpoC2,…) contain a single intron, and 11 genes (trnk-UUU, trnS-CGA, ycf3,…) have two introns.
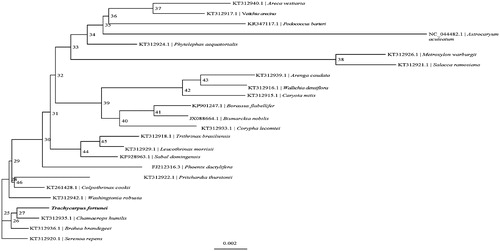
Phylogenetic analysis suggested that T. fortunei is closely clustered with Chamaerops humilis (), which was generated based on the 24 complete cp genomes. The sequences were initially aligned using MAFFT (Katoh and Standley Citation2013). The phylogenetic tree was built using IQ-TREE (Nguyen et al. Citation2015) with 1000 bootstrap. The result shows a foundation for chloroplast genome engineering of T. fortunei in the future.
Figure 1. Phylogenetic relationships of 24 species based on the maximum-likelihood (ML) analysis of T. fortunei.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
The data that support the findings of this study are openly available in GenBank of NCBI at http://www.ncbi.nim.nih.gov, reference number MT712077.
Additional information
Funding
References
- Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD. 2012. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 19(5):455–477.
- Coil D, Jospin G, Darling AE. 2015. A5-miseq: an updated pipeline to assemble microbial genomes from Illumina MiSeq data. Bioinformatics. 31(4):587–589.
- Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 30(4):772–780.
- Nguyen LT, Schmidt HA, Von Haeseler A, Minh BQ. 2015. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. 32(1):268–274.
- Shi L, Chen H, Jiang M, Wang L, Wu X, Huang L, Liu C. 2019. CPGAVAS2, an integrated plastome sequence annotator and analyzer. Nucleic Acids Res. 47(W1):W65–W73.
- Yunfa D. 2005. The many uses and exploitation of Trachycarpus fortunei China. Wild Plant Resour. 24:25–27.