
Abstract
Bambusa beecheyana var. pubescens is mainly distributed in South China to Southwest China, growing on hillsides or river banks. In the current study, we sequenced the complete chloroplast genome of B.beecheyana var. pubescens and reported for the first time. The genome is 139,402 bp in total length, include a large single-copy (LSC) region of 82,936 bp, small single-copy (SSC) region of 12,868 bp, a pair of invert repeats (IR) regions of 21,799 bp. Plastid genome contains 132 genes, 85 protein-coding genes, 39 tRNA genes, and 8 rRNA genes. Phylogenetic analysis based on 25 chloroplast genomes indicates that B. beecheyana var. pubescens is closely related to Bambusa oldhamii, Bambusa ventricosa and Bambusa ventricosa multiplex in Bambusodae.
Bambusa beecheyana var. pubescens (http://www.theplantlist.org/) is one of the taller bamboo species with the developed and simple planting method to establish. It is an outstanding bamboo species for landscaping and soil and water conservation. The chloroplasts (cp) genome has a maternal inheritance and conserved structure, that has been used to examine the developmental and phylogenetic relationships of plants. (Wang et al. Citation2018).Therefore, we reported the complete cp genomeof B. beecheyana var. pubescens based on Illumina pair-end sequencing data. Fresh leaves tissues of B. beecheyana var. pubescens were collected from Fujian province, China (Fujian Agriculture and Forestry University, Bamboo Garden, Fuzhou:119°14′16″E, 26°5′7″N), and dried into silica gel instantaneously. The specimens were preserved in the Herbarium of College of Forestry, Fujian Agriculture and Forestry University (specimen code HTY021). DNA was extracted from fresh leaves tissues, while its quantification was verified using Agarose gel electrophoresis and concentration Nanodrop, with 500 bp randomly interrupted sequence by the Covaris ultrasonic breaker for library construction. Approximately, 2.0 GB of raw data were generated with 150 bp paired-end read lengths. The Illumina High-throughput sequencing platform (HiSeq2500) data were filtered by the script in the NOVOPlasty (Dierckxsens et al. Citation2017).The complete plastid genome of Salix rehderiana (GeneBank accession: MG262367) as reference and plastid genome of B. beecheyana var. pubescens were assembled by GetOrganelle pipe-line (https://github.com/Kinggerm/GetOrganelle), it can get the plastid-like reads, and the reads were viewed and edited by Bandage (Wick et al. Citation2015). The cp genome annotation was assembled based on the comparison by Geneious v 11.1.5 (Biomatters Ltd, Auckland, New Zealand) (Kearse et al. Citation2012). The annotation results were drawn with the online tool OGDRAW (http://ogdraw.mpimp-golm.mpg.de/) (Lohse et al. Citation2013).
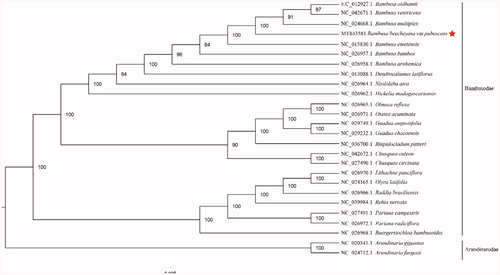
The complete cp genome of B. beecheyana var. pubescens (GenBank accession: MT843581) was 1,39,402 bp in length, with a large single-copy (LSC) region of 82,936 bp, a small single-copy (SSC) region of 12,868 bp, and a pair of inverted repeats (IR) regions of 21,799 bp. The complete cp genome consisted of 132 genes, having 85 protein-coding genes, 39 tRNA genes, and 8 rRNA genes. The complete cp genome GC content was 38.92%. In order to reveal the phylogenetic position of B. beecheyana var. pubescens with other members of Bambusodae, we performed a phylogenetic analysis based on 23 complete cp genomes of Bambusodae, and two taxa (Arundinaria gigantea、Arundinaria fargesii) as outgroups. All of them were downloaded from NCBI GenBank. The sequences were aligned by MAFFT v7.307 (Katoh and Standley Citation2013), and the phylogenetic tree was constructed by RAxML (Stamatakis Citation2014). The phylogenetic tree revealed that B. beecheyana var. pubescens was most closely related to B. oldhamii, B. ventricosa and B.multiplex with strong support ().
Figure 1. Phylogenetic analysis of 23 species of Bambusodae and two taxa (Arundinaria gigantea, Arundinaria fargesii) as outgroup based on plastid genome sequences by RAxML, bootstrap support value near the branch.
Acknowledgment
The author thanks to anonymous reviewers for their helpful suggestions and critical comments on this manuscript.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
The data that support the findings of this study are openly available at https://www.ncbi.nlm.nih.gov/ GeneBank with following accession number; MT843581 (BankIt 2370800 HTY021).
Additional information
Funding
References
- Dierckxsens N, Mardulyn P, Smits G. 2017. NOVOPlasty: de novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 45(4):e18.
- Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 30(4):772–780.
- Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, et al. 2012. Geneious basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 28(12):1647–1649.
- Lohse M, Drechsel O, Kahlau S, Bock R. 2013. OrganellarGenomeDRAW-a suite of tools for generating physical maps of plastid and mitochondrial genomes and visualizing expression data sets. Nucleic Acids Res. 41(Web Server issue):W575–W581.
- Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 30(9):1312–1313.
- Wang J, Li C, Yan C, Zhao X, Shan S. 2018. A comparative analysis of the complete chloroplast genome sequences of four peanut botanical varieties. PeerJ. 6:e5349.
- Wick RR, Schultz MB, Zobel J, Holt KE. 2015. Bandage: interactive visualization of de novo genome assemblies. Bioinformatics. 31(20):3350–3352.