
Abstract
The complete mitochondrial genome (mtDNA) of Microhyla beilunensis (Anura: Microhylidae) was sequenced and annotated. The length of mtDNA sequences of M. beilunensis was 16,721 bp, and encoded 13 protein-coding genes (PCGs), 22 transfer RNA (tRNA) genes, 2 ribosomal RNA (rRNA) genes, and a control region. The overall nucleotide composition of this genome was 29.1% A, 24.5% C, 14.5% G, 31.9% T, with a total A + T content of 61%. Phylogenetic analysis using Bayesian Inference (BI) method revealed that M. beilunensis was closely related with other 8 species from the genus Microhyla. The mtDNA dataset could be utilized for studying the molecular ecology and population genetics of Microhylid frogs.
Microhyla beilunensis (Anura: Microhylidae) is an endemic species in East China, which is mainly distributed in Ningbo and Lishui cities, Zhejiang Province (Zhang et al. Citation2018; Chen et al. Citation2020), and its distribution range is overlapped with other Microhyla species (e.g., M. fissipes, M. heymonsi) (Chen et al. Citation2020). The mitochondrial genome (mtDNA) dataset as the genetic markers could be used for studying the molecular ecology and population genetics of amphibians. In this study, we sequenced the mitochondrial genome of M. beilunensis, representing the ninth mtDNA from the genus Microhyla.
The sample (LSU20200425001JL) of M. beilunensis was collected in Jiulongshan National Nature Reserve (28.37°N, 118.90°E), Zhejiang, China, and stored at −80 °C in our laboratory at Lishui University. Total genomic DNA was isolated using EasyPure Genomic DNA Kit (TransGen Biotech Co, Beijing, China). The mtDNA sequences of M. beilunensis were obtained by next-generation sequencing (Illumina NovaSeq 6000; Novogene Bioinformatics Technology Co. Ltd., Tianjin, China) for PE 2 × 150 BP sequencing. Raw sequence data (17.57 Gb) were deposited in the Sequence Read Archive (SRA) database (accession: SRR11915114), and clean data without sequencing adapters were de novo assembled using the NOVO Plasty 3.7 (Dierckxsens et al. Citation2017).
The complete and circular mtDNA sequence is 16,721 bp in size, and has been deposited in the Genbank (accession: MT559308). The overall nucleotide composition of this genome was 29.1% A, 24.5% C, 14.5% G, 31.9% T, with a total A + T content of 61%. It contained a common set of 37 mitochondrial genes including 13 protein-coding genes (PCGs), 22 transfer RNA (tRNA) genes, 2 ribosomal RNA (12S rRNA and 16S rRNA) genes, and 2 non-coding regions of an L-strand replication origin and a D-loop region. Within 37 mitochondrial genes, most genes including 12 PCGs, 14 tRNA genes and 2 rRNA genes were encoded by the H-strand, while eight tRNAs genes and ND6 gene were encoded by the L-strand. The gene arrangement pattern and transcription directions were identical to previous studies in the genus Microhyla (Yong et al. Citation2016; Zhao et al. Citation2018). The total length of 13 PCGs is 11,349 bp and most PCGs start with typical ATK codons except COI with GTG; the AGG was found as the stop codon in ND1, ND5 and COI, the TAG was found as the stop codon in ND2, the AGA was found as the stop codon in ND6, the TAA was found as the stop codon in ATP8, ND3, ND4L, Cytb, while 3 PCGs (COII, COIII and ND4) stop with a single T, and ATP6 stop with TA, which were presumably completed as TAA by post-transcriptional polyadenylation (Anderson et al. Citation1981). The 22 tRNA genes varied in size from 66 to 74 bp. The 2 rRNA genes were 934 bp (12S) and 1583 bp (16S), respectively.
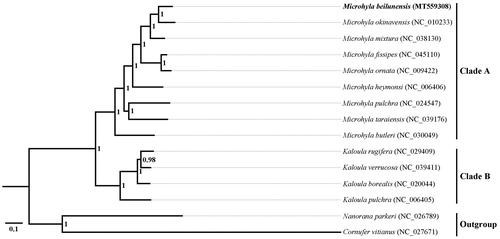
We used the Bayesian Inference method to reconstruct the phylogeny of 13 Microhylid species including M. beilunensis based on 13 PCGs (11,427 bp). Cornufer vitianus (Anura: Ceratobatrachidae) and Nanorana parkeri (Anura: Dicroglossidae) were selected as the outgroups. The best-fit substitution model (GTR + I + G) was selected by MrModelTest 2.3 under the corrected Akaike’s Information Criterion. As shown in , M. beilunensis was positioned near M. okinavensis within the genus Microhyla (Clade A). However, the results from Gorin et al. (Citation2020) showed that the phylogenetic relationship between M. beilunensis and M. mixtura was closer than that between M. beilunensis and M. okinavensis based on 3207 bp of combined mtDNA and nuDNA data. Therefore, more molecular evidence is needed to determine the evolutionary relationship among these three species in the future. In additaion, the genus Microhyla was clustered with the genus Kaloula (Clade B) as the sister genus, which is similar to previous results (Yong et al. Citation2016; Zhao et al. Citation2018). It indicated that our new determined mtDNA sequences could be utilized for studying the molecular ecology and population genetics of Microhylid frogs.
Figure 1. Phylogenetic relationships of Microhyla beilunensis and other 12 Microhylid species based on 13 concatenated mitochondrial PCGs were analyzed with Bayesian inference (BI) method. Numbers on node are posterior probability.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
The data that support the findings of this study are openly available in NCBI at www.ncbi.nlm.nih.gov [MT559308].
Additional information
Funding
References
- Anderson S, Bankier AT, Barrell BG, de Bruijn MH, Coulson AR, Drouin J, Eperon IC, Nierlich DP, Roe BA, Sanger F, et al. 1981. Sequence and organization of the human mitochondrial genome. Nature. 290(5806):457–465.
- Chen ZQ, Lin YF, Tang Y, Ding GH, Wu YQ, Lin ZH. 2020. Acoustic divergence in advertisement calls among three sympatric Microhyla species from East China. PeerJ. 8:e8708.
- Dierckxsens N, Mardulyn P, Smits G. 2017. NOVOPlasty: de novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 45(4):e18.
- Gorin VA, Solovyeva EN, Hasan M, Okamiya H, Karunarathna DMSS, Pawangkhanant P, de Silva A, Juthong W, Milto KD, Nguyen LT, et al. 2020. A little frog leaps a long way: compounded colonizations of the Indian Subcontinent discovered in the tiny Oriental frog genus Microhyla (Amphibia: Microhylidae). PeerJ. 8:e9411.
- Yong HS, Song SL, Lim PE, Eamsobhana P, Tan J. 2016. Complete mitochondrial genome and phylogeny of Microhyla butleri (Amphibia: Anura: Microhylidae). Biochem Syst Ecol. 66:243–253.
- Zhang MH, Fei L, Ye CY, Wang WF, Wang B, Jiang JP. 2018. A new species of genus Microhyla (Amphibia: Anura: Microhylidae) from Zhejiang Province. China. Asian Herpetol Res. 9:135–148.
- Zhao YY, Meng HZ, Su LN. 2018. The complete mitochondrial genome of the mixtured pygmy frog Microhyla mixtura (Anura, Microhylidae). Conservation Genet Resour. 10(3):427–430.