
Abstract
The black-winged fly, Felderimyia fuscipennis (Diptera: Tephritidae), is an insect pest of bamboo shoot, mainly distributed in Thailand, Malaysia and Yunnan Province and Guangxi Autonomous Region, China. The complete sequence of the mitogenome of F. fuscipennis has been determined in this study. The whole mitogenome sequence is 16,536 bp in length, which totally contains 13 protein-coding genes (PCGs), 2 rRNA genes, 22 tRNA genes, and a non-coding region (putative control region, CR). The phylogeny indicates that F. fuscipennis of subfamily Trypetinae was monophyletic and clearly separated from both Dacinae and Tephritinae with high bootstrap value supported.
The black-winged fly, Felderimyia fuscipennis, is an insect pest of bamboo shoot, belongs to the genus Felderimyia of the family Tephritidae (Diptera) (Wang and Chen Citation2002). The shoots of various species of bamboo serve as hosts for larvae of F. fuscipennis (Surakrai Citation2005). The fly is mainly distributed in Thailand, Malaysia, Yunnan Province and Guangxi Autonomous Region, China (Yu et al. Citation2010). In this study, we reported complete mitogenome of the F. fuscipennis from Bama, China, and it is the first mitogenome for the genus Felderimyia. The results can provide valuable information to greatly facilitate phylogenetics, population genetics, and species identification between the species of family Tephritidae.
The samples for adult flies were captured by net from Bama (25.15°N, 107.25°E), China, on 17 June 2019. Specimens were deposited in the museum of Southwest Forestry University (Voucher FFBM20190617), Kunming, China. Genomic DNA was extracted according to the manufacturer’s instruction in the DNeasy Blood and Tissue kit (Qiagen, Hilden, Germany) and sequenced on Illumina’s HiSeq2500 platform (Illumina, San Diego, CA, USA). The clean data were then assembled using the NOVOPlasty software (Dierckxsens et al. Citation2017). The genes annotation in the mitogenome was doing with SPAdes v.3.9.0 (Bankevich et al. Citation2012) and some genes were annotated manually by aligning with the mitochondrial genomes of relatively related species. Both protein-coding genes (PCGs) and rRNA genes were predicted by using MITOS tools (Bernt et al. Citation2013), and the tRNA of mitogenome was identified through tRNAscan-SE webserver (Lowe and Chan Citation2016).
The complete mitogenome of F. fuscipennis is a closed circular double-strand DNA molecule and 16,536 bp in length (GenBank accession number: MT702879). The base pairs composition is 38. 2% for A, 17.5% for C, 10.3% for G, and 34.0% for T. The sequence shows weakly positive AT-skew (0.0582) and negative GC-skew (-0.2593). The whole mitogenome encodes the classical 37 genes of invertebrate, which includes 13 protein-coding genes (PCGs), 2 rRNA genes, 22 tRNA genes, and a non-coding region (putative control region, CR). There were 23 genes (NAD2-3, NAD6, COX1-3, CYTB, ATP6, ATP8, and 14 tRNAs) encoded on the J-strand, and the remaining 14 genes (NAD1, NAD4, NAD4l, NAD5, 8 tRNAs, and 2 rRNAs) were encoded on the N-strand. The gene arrangement is the same as the most common type of the putative ancestor of insects (Boore Citation1999; Cameron Citation2014). The length of 22 tRNAs range from 62 to 72 bp. The secondary structure prediction of tRNA indicates that 21 tRNA were standard clover structure except for Serine, which is short of DHU arm. 16S rRNA is 1358 bp in length and 12S rRNA is 797 bp in length. The control region is 1633 bp in length, and has rich A + T content (83.7%).
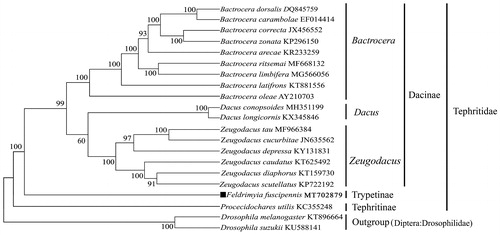
To validate the position of this species in the phylogenetic tree, a total of 21 complete mitogenomes were used in phylogenetic analysis, including 19 species in Tephritidae and 2 outgroups species from Drosophilidae, were clustered together to construct maximum likelihood tree (ML) based on the General Time Reversible model in MEGA X software (Kumar et al. Citation2018). The phylogeny indicates that F. fuscipennis in subfamily Trypetinae was clearly separated from subfamily Dacinae and subfamily Trypetinae. In subfamily Dacinae, genus Dacus and genus Zeugodacus formed a sister clade to genus Bactrocera with high bootstrap values (). Furthermore, three subfamilies were correctly identified as assigned Dacinae, Trypetinae and Tephritinae (). In conclusion, the complete mitogenome of F. fuscipennis reported in the study can provide important DNA molecular data for further phylogenetic and evolutionary analysis for family Tephritidae of order Diptera.
Figure 1. Molecular phylogeny based on complete mitogenome of the related 19 species in family Tephritidae and 2 outgroups. Tree was constructed by maximum likelihood method with 500 bootstrap replicates. Genbank accession numbers lie after the scientific name of species. The position of F. fuscipennis is marked with solid square shape.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
The data that support the findings of this study are openly available in GenBank of NCBI at https://www.ncbi.nlm.nih.gov, reference number MT702879.
Additional information
Funding
References
- Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, et al. 2012. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 19(5):455–477.
- Bernt M, Donath A, Juhling F, Externbrink F, Florentz C, Fritzsch G, Putz J, Middendorf M, Stadler PF. 2013. MITOS: improved de novo metazoan mitochondrial genome annotation. Mol Phylogenet Evol. 69(2):313–319.
- Boore JL. 1999. Animal mitochondrial genomes. Nucleic Acids Res. 27(8):1767–1780.
- Cameron SL. 2014. Insect mitochondrial genomics: implications for evolution and phylogeny. Annu Rev Entomol. 59:95–117.
- Dierckxsens N, Mardulyn P, Smits G. 2017. NOVOPlasty: de novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 45(4):e18.
- Kumar S, Stecher G, Li M, Knyaz C, Tamura K. 2018. MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol. 35(6):1547–1549.
- Lowe TM, Chan PP. 2016. tRNAscan-SE on-line: search and contextual analysis of transfer RNA genes. Nucl Acids Res. 44:54–57.
- Surakrai P. 2005. Bamboo-shoot fruit flies (Diptera: Tephritidae) of southern Thailand. Songklanakarin J Sci Technol. 27(2).
- Wang XJ, Chen XL. 2002. Taxonomic Revision of the Genus Felderimyia Hendel (Diptera: Tephritidae: Trypetinae). Acta Zootaxonomica Sinica. 27(4):802–806.
- Yu H, Deng Y-l, Wan-Zhong HE, Zhong-Shan LIU, Ding YANG, Nai-Zhong CHEN. 2010. A new record of the tephritidae(diptera) from yunnan, china. Entomotaxonomia. 32(4):289–292.