
Abstract
Sipalus gigas is the main pine-hole borer of Pinus. The length of the complete mitochondria genome of S. gigas was 17,120 bp with 33.6% GC content, there were 35 genes including 13 protein-coding genes (CDS), 20 transfer RNA genes (tRNAs), and two ribosomal RNA genes (rRNAs). This study provides useful genetic information for subsequent studying the prevention of S. gigas.
Sipalus gigas belongs to Curculionidae of Coleoptera, which is mainly distributed in Asian countries such as China, North Korea, and Japan (Furuta Citation1972). Sipalus gigas is one of the major pin-hole borers of Pinus, which including Pinus massoniana, P. koraiensis, and P. tabulaeformis (Yu et al. Citation2016). It has been seriously threatened the growth of pine trees and the processing of wood. Therefore, effective control of S. gigas is a urgent issue, while it is difficult to control S. gigas in production. Nowadays, biological control with bacteria toxins or fungi has been an important method to control insects. However, the genome of S. gigas have yet to be clearly determined. In order to provide useful genetic information for subsequent study of the prevention, the complete mitochondria genome of S. gigas was sequenced and assembled, as well as constructed phylogenies of S. gigas by Neighbor-Joining to understand the evolution relationship.
The S. gigas adults were collected from Lianjiang, Fujian Province, China (119° 38′25″E, 26°9′21″N) by the traps with sexual attractants. The specimens were stored in the Fujian Agriculture and Forestry University (SLX-202007).The total DNA was extracted from the legs of S. gigas by TruSeq DNA sample Preparation kit (Vanzyme, CHN) and purified by QIAquick Gel Extraction kit (Qiagen, GER). The mitochondrial genome was sequenced through Illumina Hiseq 2500 by Genesky Biotechnologies Inc. (Shanghai, China). In all, 566,589,34 clean reads were obtained through quality analysis and filtration. Then these clean reads were assembled by using de novo and the MITOS web server (Bernt et al. Citation2013; Hahn et al. Citation2013). And tRNA genes were predicted using tRNAscan (Lowe and Eddy Citation1997). The complete mitochondria genome length of S. gigas was 17,120 bp (GenBank accession no. MT809476). The GC content of the complete genome was 33.60% and there were 35 genes including 13 protein coding sequences, 20 tRNAs, and two rRNAs.
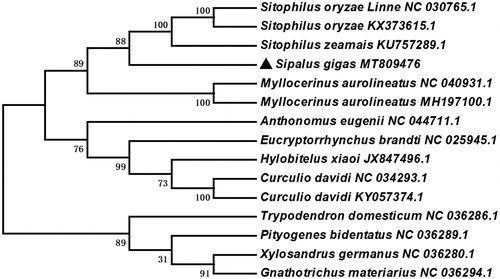
To further investigate the phylogenetic position of S. gigas, according to the genome sequence of S. gigas, the phylogenetic analysis was constructed with fourteen different species of Coleoptera by MEGA 6.0 (Tamura et al. Citation2013) using Neighbor-Joining tree model with 1000 bootstrap replicates. The Neighbor-Joining tree showed that S. gigas was closely related to Sitophilus zeamais and sister to Myllocerinus aurolineatus (). The complete mitochondrial genome of S. gigas will provide useful genetic information to better understand the genetic evolution in Sipalus, as well as in Curculionidae and other insects of Coleoptera.
Figure 1. Neighbor-Joining tree of the Sipalus gigas and related 14 different species of Coleoptera based on the genome sequence. Numbers labeled on the branch are bootstrap values.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
The data that support the findings of this study are openly available in GenBank of NCBI at https://www.ncbi.nlm.nih.gov, reference number MT809476.
Additional information
Funding
References
- Bernt M, Donath A, Jühling F, Externbrink F, Florentz C, Fritzsch G, Pütz J, Middendorf M, Stadler PF. 2013. MITOS: improved de novo metazoan mitochondrial genome annotation. Mol Phylogenet Evol. 69(2):313–319.
- Furuta K. 1972. The change of the distribution pattern of the large weevil, Hyposipalus gigas Fabricius (Coleoptera, Rhynchophoridae) within a single generation. A preliminary note. Res Popul Ecol. 13(2):216–221.
- Hahn C, Bachmann L, Chevreux B. 2013. Reconstructing mitochondrial genomes directly from genomic next-generation sequencing reads—a baiting and iterative mapping approach. Nucleic Acids Res. 41(13):e129–e129.
- Lowe TM, Eddy SR. 1997. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25(5):955–964.
- Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. 2013. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol. 30(12):2725–2729.
- Yu Z, Zhi-Tao L, Sheng-Nan Z, Yuan-Yuan Z, Chao-Hui S. 2016. Population dynamic monitoring of Hyposipalus gigas Linnaeus in Conghua. J Heb For Sci Technol. 2016(3):24–25.