
Abstract
In this study, the first complete mitochondrial genome of Chrysolina aeruginosa Fald was assembled and analyzed. The total length of this mitochondrial genome is 16,335 base pairs. It consists of 13 protein-coding genes, 22 transfer RNAs, two ribosomal RNAs, and an AT-rich region. Phylogenomic analysis indicated that C. aeruginosa Fald is sister to Chrysodinopsis sp. This study provides new molecular data for the further taxonomic and phylogenetic studies of the Chrysomelidae of Coleoptera.
Chrysolina aeruginosa Fald is a major pest of Artemisia ordosica Krasch. In recent years, this phytophagous beetle has spread rapidly throughout northwest China, leading to mass mortalities of A. ordosica Krasch, which has produced great damage to the local ecology (Da-Zhi et al. Citation2014). In the present study, the complete mitochondrial genome of C. aeruginosa Fald is reported contributing for better understanding its evolution and population genetics, and also providing significant information for the phylogeny of Chrysomelidae.
The samples of C. aeruginosa Fald were caught by the traps of attractive substance from Minhou, Fujian Province, China (119°4′35″E, 26°14′9″N). All voucher specimens were assigned with a unique code and deposited in the Key Laboratory of Integrated Pest Management in Ecological Forests, Fujian Province University, Fujian Agriculture and Forestry University (voucher no.YJ-202006). The genomic DNA was extracted using a phenolchloroform extraction protocol from the legs of samples (Russell and Sambrook Citation2001). DNA quality and concentration were determined using Nanodrop (Thermo Fisher Scientific,Waltham, MA, USA). Then, the DNA library with fragments of 300 bp in size was constructed by PCR amplification and the size selection by Agencourt AMPure XP-PCR Purification Beads (Beckman Coulter, CA, USA) and Agencourt SPRIselect Beads (Beckman Coulter, CA, USA). The multiple samples were mixed and sequenced using Illumina Hiseq 2500 (Genesky Biotechnologies Inc. Shanghai, China).
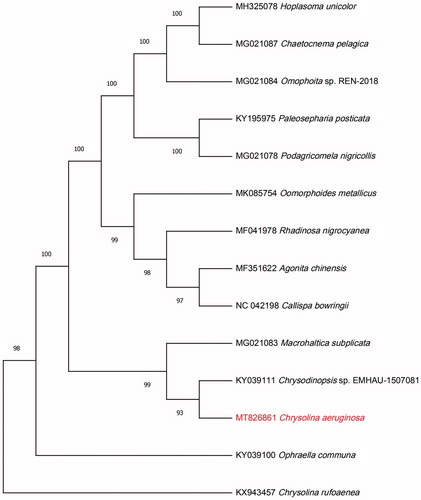
A total of 46,513,652 clean reads were obtained by filtration from the 47,161,712 raw reads. After the de novo assembly of metaSPAdes (Nurk et al. Citation2017), the complete circular mitogenome was obtained. The total length of this mitochondrial genome is 16,335 base pairs (GenBank accession No: MT826861). Three characteristics of protein-coding sequence, tRNA, and rRNA were obtained by the annotated result of mitoMaker (Bernt et al. Citation2013). And, tRNA genes were predicted by using tRNAscan (Lowe and Eddy Citation1997). A total of 37 genes were annotated, which contained 13 protein-coding genes, 22 transfer RNA (tRNA), and two ribosomal RNA (rRNA), there was also an AT-rich region. To confirm the phylogeny of C. aeruginosa Fald, 13 complete genomes of different species of Chrysomelidae were obtained from GenBank, and aligned using HomBlocks software (Bi et al.Citation2018). A Maximum Likelihood (ML) tree with 1000 bootstrap replicates was inferred using MEGA 7.0 (Sudhir et al. Citation2016). The result of ML phylogenetic tree showed that C. aeruginosa Fald is closely related to Chrysodinopsis sp. EMHAU-1507081(). The mitochondrial genome of C. aeruginosa Fald will provide useful genetic information for further study on genetic diversity and genetic evolution of Chrysomelidae species.
Figure 1. The Maximum Likelihood (ML) phylogenetic tree based on 14 mitochondrial genome of Chrysomelidae. Values along branches correspond to ML bootstrap percentages.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
The data that support the findings of this study are openly available in”NCBI”at https://www.ncbi.nlm.nih.gov/, reference number MT826861.
Additional information
Funding
References
- Bernt M, Donath A, Juhling F, Externbrink F, Florentz C, Fritzsch G, Pütz J, Middendorf M, Stadler PF. 2013. MITOS: improved de novo metazoan mitochondrial genome annotation. Mol Phylogenet Evol. 69(2):313–319.
- Bi G, Mao Y, Xing Q, Cao M. 2018. HomBlocks: a multiple-alignment construction pipeline for organelle phylogenomics based on locally collinear block searching. Genomics. 110(1):18–22.
- Da-Zhi Z, Na Z, Yue-Cheng LI, et al. 2014. Population dynamics of Chrysolina aeruginosa in Ningdong region of Ningxia and the related influencing factors. Chinese J Ecol. 32(10):346–351.
- Lowe TM, Eddy SR. 1997. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25(5):955–964.
- Nurk S, Meleshko D, Korobeynikov A, Pevzner PA. 2017. metaSPAdes: a new versatile metagenomic assembler. Genome Res. 27(5):824–834.
- Russell DW, Sambrook J. 2001. Molecular cloning: a laboratory manual. 1(6). New York: Cold Spring Harbor Laboratory Press; p. 1–28.
- Sudhir K, Glen S, Koichiro T. 2016. MEGA7: molecular evolutionary geneticsanalysis version 7.0 for bigger datasets. Mol Biol E. 33(7):1870–1874.