
Abstract
The complete mitochondrial (mt) genomes of five subspecies of the Eurasian (Common) magpie Pica pica were determined for the first time. Lengths of the circular genomes comprise 13 protein-coding genes, two rRNA genes (for 12S rRNA and 16S rRNA), 22 tRNA genes, and the non-coding control region (CR). Gene content and lengths of the genomes (16,936–16,945 bp) are similar to typical vertebrate mt genomes. The subspecies studied differs by several single substitutions and indels, especially in the CR. The phylogenetic tree based on complete mt genomes shows a deep divergence of the two groups of subspecies which supports the proposed division into two distinct species: P. pica and P. serica.
Complete mitochondrial (mt) genomes are still rarely explored in intraspecies systematics. Taxonomy, species borders, and intraspecies structure of the Eurasian (Common) magpie Pica pica L. (Passeriformes, Corvidae) are a matter of debate. It is widespread in the Holarctic. For the Palearctic taxa, taxonomy varies from accepting a single species with 12 subspecies (Madge and Burn Citation1999; Kryukov et al. Citation2017) to division into four species (Song et al. Citation2018). Only one complete mt genome of P. pica has been reported previously (GenBank HQ915867). Here, we describe the complete mt genomes of five subspecies (sensu Kryukov et al. Citation2017) of magpie and use them for assessing intraspecies relationships.
Tissue (blood or feather) samples were collected: P. p. camtschatica in Kamchatka Peninsula, Russia (54.30N, 156.00E); P. p. jankowskii in Primorsky Krai, Russia (43.84N, 131.86E); P. p. fennorum in Moscow region, Russia (55.75N, 38.01E); P. p. leucoptera in Mongolia (48.45N, 115.35E); P. p. melanotos in Buitrago del Lozoya, Spain (40.99N, 3.64W). The samples are stored in the tissue and DNA collections of FSCEATB, Russia (#2320 for camtschatica, #2205 for jankowskii, #1215 for fennorum, and #2385 for leucoptera), and MNCN, Spain (#2804 for melanotos). DNA was extracted with DNeasy Blood and Tissue kit (Qiagen, Hilden, Germany). Two primer pairs were designed for the amplification of two overlapping fragments: mtPica-66F (GACAAAAGACTTAGTCCTAACCTTACTGTT) and mtPica-7010R (GTGGTTTATGCGGTTGGCTTGAA) for a ∼7-kb fragment; mtPica-5359F (CCTCTGTAAAAAGGACTACAGCCTA) and mtPica-68R (AGTAAGGTTAGGACTAAGTCTTTTG) for a ∼11-kb fragment. The amplicons obtained were individually tagged with barcode sequences and sequenced using the Oxford Nanopore MinION with Flow Cell R10 and the protocol ‘1d-native-barcoding-genomic-dna-NBE_9065_v109_revI_23May2018.’ The reads were aligned one by one to the reference genome HQ915867 with AliView v. 1.17.1 (Larsson Citation2014) and consensus sequences were completed using Consensus Tool (http://kirr.dyndns.org/bio/consensus/). Annotation was performed with MITOS (Bernt et al. Citation2013).
The assembled genomes are 16,936–16,945 bp in length and each contains 37 genes comprising 13 protein-coding genes, two rRNA genes (for 12S rRNA and 16S rRNA), and 22 tRNA genes. The control region (CR) is located between tRNA-Glu and tRNA-Phe genes and has a length of 1351–1362 bp. The H- and L-strands encode 28 and nine genes, respectively. The gene arrangement is the same in all genomes studied. GC content of the new genomes is 43.3–43.5%. Proportions of A, T, C, and G in subspecies have the following ranges (in %): 31–31.1, 25.4–25.6, 29.2–29.5, and 14.0–14.1, respectively.
For illustrating phylogenetic relations of the complete mt genomes obtained, we constructed a maximum-likelihood (ML) tree () using MEGA X (Kumar et al. Citation2018).
Figure 1. Maximum-likelihood phylogenetic tree for the five newly obtained complete mitochondrial genomes of Pica pica s.l., and the previously determined sequence of Pica pica (GenBank HQ915867). Garrulus glandarius (JN018413) is used as outgroup. Numbers at nodes are bootstrap support values in %.
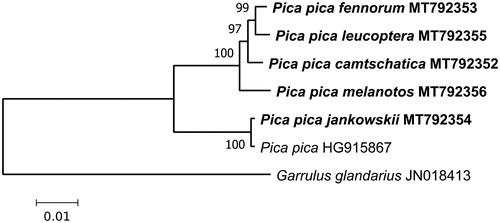
The western group of subspecies includes fennorum, melanotos, leucoptera, as well as geographically distant camtschatica. Jankowskii is nearly identical to HQ915867. The two clades are well differentiated by p-distance of .039 on average. In general, this is consistent with the previous results obtained using individual genes: cytB and CR (Kryukov et al. Citation2004, Citation2017; Haring et al. Citation2007); cytB, ND2, and two nuclear introns (Song et al. Citation2018). This degree of divergence supports classifying the two clades as separate species.
The subspecies name of HQ915867 is not mentioned in its GenBank entry. Its near identity to jankowskii suggests that it could belong to jankowskii or a closely related subspecies, such as serica or anderssoni. Kryukov et al. (Citation2017) showed close affinity of serica and jankowskii, while Song et al. (Citation2018) showed near identity of serica and anderssoni, and suggested grouping them under Pica serica species. Based on our complete mt genome data, we support classifying Pica serica as a species, distinct from P. pica (the western clade). Pica serica species may include jankowskii, serica, and anderssoni subspecies.
Continuation of genomic studies is necessary for clarifying taxonomy of the remaining subspecies.
Acknowledgements
We are grateful for Elisabeth Haring for editing the manuscript. We are thankful to Yaroslav Redkin and Eugeny Lobkov for providing samples.
Disclosure statement
No potential conflicts of interest were reported by the author(s).
Data availability statement
The data that support the findings of this study are openly available in the National Center for Biotechnology Information database (NCBI/GenBank) at https://www.ncbi.nlm.nih.gov/, reference numbers MT792352, MT792353, MT792354, MT792355, and MT792356.
Additional information
Funding
References
- Bernt M, Donath A, Jühling F, Externbrink F, Florentz C, Fritzsch G, Pütz J, Middendorf M, Stadler PF. 2013. MITOS: improved de novo Metazoan mitochondrial genome annotation. Mol Phylogenet Evol. 69(2):313–319.
- Haring E, Gamauf A, Kryukov A. 2007. Phylogeographic patterns in widespread corvid birds. Mol Phylogenet Evol. 45(3):840–862.
- Kryukov A, Iwasa MA, Kakizawa R, Suzuki H, Pinsker W, Haring E. 2004. Synchronic east–west divergence in azure-winged magpies (Cyanopica cyanus) and magpies (Pica pica). J Zool Syst Evol Res. 42(4):342–351.
- Kryukov AP, Spiridonova LN, Mori S, Arkhipov VY, Red'kin YA, Goroshko OA, Lobkov EG, Haring E. 2017. Deep phylogeographic breaks in magpie Pica pica across the Holarctic: concordance with bioacoustics and phenotypes. Zool Sci. 34(3):185–200.
- Kumar S, Stecher G, Li M, Knyaz C, Tamura K. 2018. MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol. 35(6):1547–1549.
- Larsson A. 2014. AliView: a fast and lightweight alignment viewer and editor for large data sets. Bioinformatics. 30(22):3276–3278.
- Madge S, Burn H. 1999. Crows and Jays: a guide to the Crows, Jays and Magpies of the world. London: C Helm.
- Song G, Zhang R, Alström P, Irestedt M, Cai T, Qu Y, Ericson PGP, Fjeldså J, Lei F. 2018. Complete taxon sampling of the avian genus Pica (Magpies) reveals ancient relictual populations and synchronous Late-Pleistocene demographic expansion across the Northern Hemisphere. Avian Biol. 49:e01612.