
Abstract
The complete mitogenome of Diaporthe nobilis NIE8444 (KCTC No. 56710) isolated from alpine conifer Abies nephrolepis is determined by the Illumina Hiseq4000 platform in this study. This mitogenome consists of 67,437 bp length with 31.45% G + C content. A total of 51 genes were predicted in this mitogenome: 21 protein-coding genes, 2 rRNAs and 28 tRNAs. Phylogenetic tree based on small subunit ribosomal RNA of mitochondria showed that D. nobilis was close to D. longicolla. This complete mitogenome of D. nobilis provides valuable information on the mitochondrial evolution of endophytic fungi.
Diaporthe nobilis Sacc. & Speg belongs to the class Sordariomycetes, and Diaporthe spp. are found in woody plants such as elm, pine and Korean arborvitae (Eo et al. Citation2016; Park et al. Citation2017). In this study, D. nobilis NIE8444 (KCTC No.56710) was isolated from healthy needles as endophytic fungi of Abies nephrolepis (Trautv.) Maxim, an alpine conifer and vulnerable species to climate change distributed in Mt. Nochu (37° 32′14ʺN, 128°45′36ʺE) of Korea. This fungus was deposited in the Korean Collection for Type Cultures (KCTC) and 19 isolates were successfully stored. Genomic DNA was extracted from D. nobilis NIE8444 mycelium using the Qiagen DNeasy DNA prep kit (Qiagen, Hilden, Germany). Libraries were constructed using the TruSeq Nano DNA library (Illumina Inc., San Diego, CA) and sequenced on the Illumina HiSeq4000 Platform (Illumina Inc) with 101-bp paired-end reads.
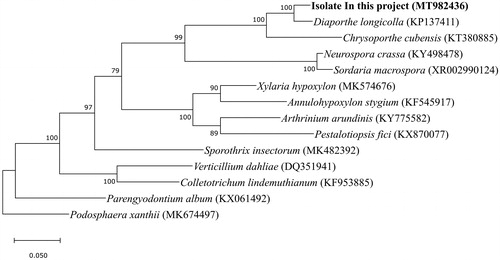
Raw reads were quality controlled using the Trimmomatic v0.36 program (Bolger et al. Citation2014). The trimmed reads were de novo assembled using the Ray v2.3.1 and SPAdes v3.11.1 programs (Boisvert et al. Citation2010; Bankevich et al. Citation2012). Potential mitochondrial contigs were extracted from assembled results by BLAST search with mitochondrial reference protein sequences of the GenBank database. Extracted contigs were manually merged and gap-closed using in-house scripts. The polished assembly was finally error-corrected by the BWA v0.7.17 and Pilon v1.23 programs using the short reads (Li Citation2013; Walker et al. Citation2014). Prediction and annotation of genes encoding functional proteins and ribosomal RNA genes was performed using the BLAST program with the mitochondrial DNA annotations of Diaporthe longicolla (GenBank: KP137411) and Chrysoporthe cubensis (GenBank: KT380885). Transfer RNA genes were predicted using ARAGORN v1.2.38 and tRNAscan-SE v2.0.5 (Laslett and Canback Citation2004; Schattner et al. Citation2005). For the construction of the phylogenetic tree with mitochondrial small subunit ribosomal RNA, 13 sequences retrieved from the GenBank database were used. Nucleotide sequences were aligned using the MUSCLE algorithm in MEGA X (Kumar et al. Citation2018). Tree building was performed using the maximum likelihood method based on the Tamura–Nei model (Tamura and Nei Citation1993) (). This fungal mitogenome consists of a circular chromosome of 67,437 bp length with 31.45% G + C content (GenBank accession number: MT982436). A total of 51 genes were predicted in this mitochondrial genome, 21 of which are protein-coding genes. All genes are encoded on the positive strand. Most of the protein-coding genes were the conserved mitochondrial proteins for oxidative phosphorylation containing cox1, cox2, cox3, nad1, nad2, nad3, nad4, nad4L, nad5, nad6, atp6, atp8 and atp9. The genome also has 2 rRNAs (rrnS and rrnL) and 28 tRNAs.
Figure 1. Unrooted maximum likelihood tree of small subunit ribosomal RNA sequences originated from mitochondria of the class Sordariomycetes. Numbers above branches show maximum likelihood bootstrap supports from 1000 nonparametric replicates. The scale represents the number of substitutions per site. All positions containing gaps and missing data were eliminated. There were a total of 1785 positions in the final dataset.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
The data that support the findings of this study will be available in GenBank at https://www.ncbi.nlm.nih.gov/, accession number MT982436.
Additional information
Funding
References
- Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD. 2012. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 19(5):455–477.
- Boisvert S, Laviolette F, Corbeil J. 2010. Ray: simultaneous assembly of reads from a mix of high-throughput sequencing technologies. J Comput Biol. 17(11):1519–1533.
- Bolger AM, Lohse M, Usadel B. 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 30(15):2114–2120.
- Eo J-K, Lee B-H, Eom A-H. 2016. Diversity of endophytes isolated from Thuja koraiensis Nakai in the Korean Peninsula. Kor J Mycol. 44(2):113–117.
- Kumar S, Stecher G, Li M, Knyaz C, Tamura K. 2018. MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol. 35(6):1547–1549.
- Laslett D, Canback B. 2004. ARAGORN, a program to detect tRNA genes and tmRNA genes in nucleotide sequences. Nucleic Acids Res. 32(1):11–16.
- Li H. 2013. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv. 13033997. [In preprint]
- Park S, Lee S-Y, Lee J-J, Back C-G, Lee HB, Jung H-Y. 2017. First report of Diaporthe tectonae isolated from soil in Korea. Kor J Mycol. 45(1):83–89.
- Schattner P, Brooks AN, Lowe TM. 2005. The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Res. 33(Web Server issue):W686–W689.
- Tamura K, Nei M. 1993. Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol Biol Evol. 10(3):512–526.
- Walker BJ, Abeel T, Shea T, Priest M, Abouelliel A, Sakthikumar S, Cuomo CA, Zeng Q, Wortman J, Young SK, et al. 2014. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLOS One. 9(11):e112963