
Abstract
Two complete chloroplast genomes of Hippuris vulgaris (H. vulgaris_A and H. vulgaris_B), representing two distinct clades in China, were sequenced and assembled in this study. The circular genomes were 152,763 and 152,713 bp in length and exhibit a typical quadripartite structure of the large single-copy (LSC, 82,983/82,949 bp) and small single-copy (SSC, 18,294/18,278 bp) regions, separated by a pair of inverted repeats (IRs, both 25,743 bp). Both two cp genomes identically contain 133 genes, including 88 protein-coding genes, 37 tRNA, and eight rRNA genes. The phylogenetic analysis within Plantaginaceae demonstrated Hippuris an independent clade included in the expanded Plantaginaceae.
Hippuris vulgaris L., previously belonging to the monogeneric family Hippuridaceae, now is included in an expanded Plantaginaceae (Olmstead et al. Citation2001; Albach et al. Citation2005). H. vulgaris is an aquatic perennial herb with a circumboreal distribution confined in temperate, boreal, and sub-arctic regions (Wan Citation2000; Elven et al. Citation2012). In Inner Mongolia and Tibet of China, H. vulgaris is used as an endemic Chinese medicine to cure tuberculosis and cough (Shang et al. Citation2012). In this study, we firstly assembled and characterized the complete chloroplast genomes of H. vulgaris, which will provide organelle molecular basis for further research of this medicinal aquatic plant.
Fresh leaves of two individuals (H. vulgaris_A and H. vulgaris_B), representing two distinct clades revealed in previous research (Lu et al. Citation2016) were collected separately from Jiagedaqi District (124°32.556′E, 51°50.416′N) and Fuyu County (124°32.954′E, 47°44.450′N) in Heilongjiang Province of China, and dried with silica gel. The vouch specimens (No. Xu7157 and Xu 639) were deposited in Herbarium of the Wuhan University. Total genomic DNA was extracted from ∼3 mg materials using DNA Plantzol Reagent (Invitrogen, Carlsbad, CA) following the manufacturer’s protocol. Purified DNA was sheared into ∼500 bp fragments, and the paired-end sequencing libraries were constructed according to the Illumina standard protocol (Illumina, San Diego, CA). Genomic DNA of two samples was sequenced using an Illumina BGISEQ-500 (Illumina, San Diego, CA) at Beijing Genomics Institute (BGI; Shenzhen, China). Illumina paired-end sequencing generated a total of 20,176,296 and 20,662,298 bp raw reads after removing adapters. The raw reads were then used to assemble the cp genomes using NOVOPlasty version 2.7.2 (Dierckxsens et al. Citation2017), with ATP synthase alpha subunit (atpA) gene from Veronica nakaiana (GenBank accession no. NC_031153) as the seed. Chloroplast genome annotation was performed using transferring annotations in Geneious Prime (Kearse et al. Citation2012), with the cp genome of V. nakaiana (GenBank accession no. NC_031153) as the reference. Where necessary, the positions of start and stop codons and boundaries between introns and exons were manually corrected. The two annotated complete cp genomes of H. vulgaris were deposited in GenBank under the accession no. MW044609 and MW044610. Chloroplast genomes of H. vulgaris_A and H. vulgaris_B were circular DNA molecules of 152,763 and 152,713 bp in length. Both cp genomes had a typical quadripartite structure, consisting of a pair of inverted repeats (IRa and IRb: both 25,743 bp) separated by a large single-copy region (LSC: 82,983 and 82,949 bp) and a small single-copy region (SSC: 18,294 and 18,278 bp). The overall GC content was both 37.6%. The IR regions had higher GC content (43.1 and 43.2%) than the LSC (35.6 and 35.7%) and SSC (30.6 and 30.8%) regions. Both two cp genomes encoded a set of 133 genes, containing 88 protein-coding genes, 37 tRNA genes, and eight rRNA genes.
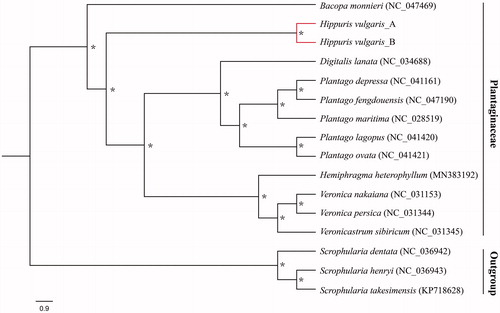
Eleven species in Plantaginaceae and three species of Scrophularia with available chloroplast genomes were selected to study the phylogenetic placement of H. vulgaris (). The sequence alignment was conducted using MAFFT version 7.3 (Katoh and Standley Citation2013). The phylogenetic tree was constructed using IQTREE version 1.6.7 (Nguyen et al. Citation2015), with the best selected GTR + F + R3 model and 5000 bootstrap replicates. Three Scrophularia species were used as outgroups. The result confirmed that H. vulgaris was included in Plantaginaceae and forms an independent clade in present data extent.
Figure 1. The best maximum likelihood (ML) phylogram inferred from 16 complete chloroplast genome sequences in Plantaginaceae and Scrophularia (accession numbers were listed and ‘*’ indicates the bootstrap support values = 100%).
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
The DNA matrix and phylogenetic tree that support the findings of this study are openly available via the DOI http://dx.doi.org/10.25833/qdhr-0c03.
Additional information
Funding
References
- Albach DC, Meudt HM, Oxelman B. 2005. Piecing together the “new” Plantaginaceae. Am J Bot. 92(2):297–315.
- Dierckxsens N, Mardulyn P, Smits G. 2017. NOVOPlasty: de novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 45(4):e18.
- Elven R, Murray DF, Solstad H. 2012. Hippuris L. In: Flora of North America Association, editors. Flora of North America. England: Oxford University Press; Vol. 17. p. 55.
- Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 30(4):772–780.
- Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, et al. 2012. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 28(12):1647–1649.
- Lu QX, Zhu JN, Yu D, Xu XW. 2016. Genetic and geographical structure of boreal plants in their southern range: phylogeography of Hippuris vulgaris in China. BMC Evol Biol. 16(1):34–34.
- Nguyen LT, Schmidt HA, von Haeseler A, Minh BQ. 2015. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. 32(1):268–274.
- Olmstead RG, Depamphilis CW, Wolfe AD, Young ND, Elisons WJ, Reeves PA. 2001. Disintegration of the Scrophulariaceae. Am J Bot. 88(2):348–361.
- Shang XF, Tao CX, Miao XL, Wang DS, Tangmuke , Dawa , Wang Y, Yang YG, Pan H. 2012. Ethno-veterinary survey of medicinal plants in Ruoergai region, Sichuan province, China. J Ethnopharmacol. 142(2):390–400.
- Wan WH. 2000. Hippuridaceae. In: Chia Jui C, editor. Flora reipublicae popularis sinicae. China: Science Press; Vol. 53(2). p. 144–147.