
Abstract
We assembled a complete chloroplast genome of a pantropical legume, Canavalia rosea (Fabaceae). The chloroplast genome was 158,059 bp in length that was composed of a 77,752 bp large single copy region, a 18,993 bp small single copy region, and a pair of 30,657 bp inverted repeats. We detected 135 genes that consisted of 90 protein-coding genes, 37 tRNA genes, eight rRNA genes, and three pseudogenes (rps16 and a pair of rpl22).
Canavalia rosea (Sw.) DC. (Fabaceae) is one of the representative species of pantropical plants with sea-drifted seeds (Takayama et al. Citation2006) that have extremely wide ranges of distribution in the tropics and sub-tropics region. Gene flow via sea-dispersal is an intriguing question to understand the formation of such unique distribution of a single plant species, and chloroplast genomes will provide appropriate markers to evaluate gene flow and genetic structure because of their maternal nature of inheritance. Complete chloroplast genomes would also provide useful information to understand the reproductive strategy of the plants. In this study, we constructed the complete chloroplast genome of C. rosea using paired-end short-read data.
We collected leaf samples from a cultivated plant in the glasshouse of Iriomote Station, Tropical Biosphere Research Center, University of the Ryukyus. The plant was grown from seeds collected from a wild population of C. rosea in Senegal (at Joal-Fadiout on 3rd December 2000) collected by TK (No. 00120304). The voucher specimen is stored in the URO herbarium, University of the Ryukyus. We extracted total DNA from silica-dried leaves using the CTAB method (Doyle and Doyle Citation1987). Genomic DNA was sequenced with Illumina Hiseq2000. We obtained 22 million 150 bp paired-end reads and removed low-quality nucleotides and reads using the Trimmomatic 0.39.0 (Bolger et al. Citation2014) with a palindrome clip threshold of 30 and a simple clip threshold of 10. We assembled a chloroplast genome using the GetOrganelles pipeline (Camacho et al. Citation2009; Bankevich et al. Citation2012; Langmead and Salzberg Citation2012; Wick et al. Citation2015; Jin et al. Citation2020) and used GeSeq in CHLOROBOX web service (Tillich et al. Citation2017) for annotation of the chloroplast genome. The chloroplast genome sequence and annotation were submitted to DDBJ (DNA Data Bank of Japan) accession number LC554221.
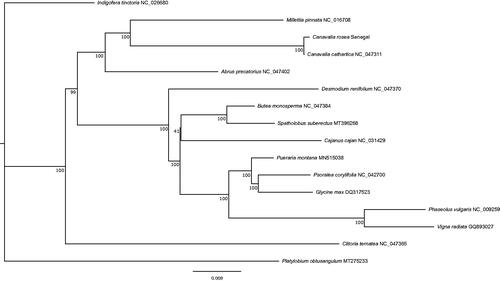
The total length of the chloroplast genome was 158,059 bp, which is 164 bp shorter than a closely related species, Canavalia cathartica Thouars, the chloroplast genome of which is available in GenBank (accession No. NC_047311). The large single copy (LSC) and small single copy (SSC) regions were 77,752 bp and 18,993 bp, respectively. The length of inverted repeats was 30,657 bp. We detected 135 genes including 90 protein-coding genes, 37 tRNA genes, and 8 rRNA genes, and the numbers of genes were the same as C. cathartica NC_047311. We also detected three pseudogenes (rps16 and a pair of rpl22). Pseudogenes of rps16 were found in other individuals of C. rosea (unpublished data), which suggests possible allelic gene loss in this species. Pseudogenes of rpl22 were also reported in other legumes (Gantt et al. Citation1991). We constructed a phylogenetic tree of Millettioid/Phaseoloid clade based on the method of Zhang et al. (Citation2020) by using 84 coding regions of 15 chloroplast genomes obtained from GenBank. Each gene was aligned by MAFFT 7.4 (Katoh et al. Citation2002, Citation2005; Katoh and Toh Citation2007), then all genes were concatenated by using SeqKit (Shen et al. Citation2016). A phylogenetic tree obtained by RAxML-NG (Kozlov et al. Citation2019) with the GTR + G + I model and bootstrap was drawn using Figtree 1.4.2 (Rambaut Citation2012) 500 times. The phylogenetic tree of C. rosea chloroplast genome formed a clade with C. cathartica, which is consistent with that of Zhang et al. (Citation2020) and the generic relationships of the phylogenetic tree were consistent with that of Zhang et al. Citation2020 ().
Figure 1. The maximum-likelihood tree of Millettioid/Phaseoloid based on 84 genes in chloroplast genomes. Bootstrap values were shown under branches.
Acknowledgment
We thank Dr. A. Tidiane Ba (Université Cheikh Anta Diop de Dakar) for supporting us to collect the seed samples used in this study. We also thank Ms. H. Tsutsumi and Mr. S. Imura and K. Ishigaki for the cultivation of live plants at a glasshouse.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
Data that support the findings of this study are openly available in DDBJ (DNA Data Bank of Japan) and can be accessed at http://getentry.ddbj.nig.ac.jp/top-e.html; accession number LC554221.
Additional information
Funding
References
- Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, et al. 2012. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 19(5):455–477.
- Bolger AM, Lohse M, Usadel B. 2014. Trimmomatic: a flexible trimmer for illumina sequence data. Bioinformatics. 30(15):2114–2120.
- Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, Madden TL. 2009. BLAST+: architecture and applications. BMC Bioinf. 10(1):421.
- Doyle JJ, Doyle JL. 1987. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem Bull 19(1): 11–15.
- Gantt JS, Baldauf SL, Calie PJ, Weeden NF, Palmer JD. 1991. Transfer of Rpl22 to the nucleus greatly preceded its loss from the chloroplast and involved the gain of an intron. The EMBO J. 10(10):3073–3078.
- Jin J-J, Yu W-B, Yang J-B, Song Y, DePamphilis CW, Yi T-S, Li D-Z. 2020. GetOrganelle: a fast and versatile toolkit for accurate de novo assembly of organelle genomes.Genome Biol21, 241. https://doi.org/10.1186/s13059-020-02154-5.
- Katoh K, Toh H. 2007. PartTree: an algorithm to build an approximate tree from a large number of unaligned sequences. Bioinformatics. 23(3):372–374.
- Katoh K, Ichi Kuma K, Toh H, Miyata T. 2005. MAFFT version 5: improvement in accuracy of multiple sequence alignment. Nucleic Acids Res. 33(2):511–518.
- Katoh K, Misawa K, Kuma KI, Miyata T. 2002. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 30(14):3059–3066.
- Kozlov AM, Darriba D, Flouri T, Morel B, Stamatakis A, Wren J. 2019. RAxML-NG: a fast, scalable and user-friendly tool for maximum likelihood phylogenetic inference. Bioinformatics. 35(21):4453–4455.
- Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with bowtie 2. Nat Methods. 9(4):357–359.
- Rambaut A. 2012. FigTree v1. 4. 2, A Graphical Viewer of Phylogenetic Trees. Available from 〈http://tree.bio.ed.ac.uk/software/figtree/〉
- Shen W, Le S, Li Y, Hu F. 2016. SeqKit: a cross-platform and ultrafast toolkit for FASTA/Q file manipulation. PLoS One. 11(10):e0163962.
- Takayama K, Kajita T, Murata J, Tateishi Y. 2006. Phylogeography and genetic structure of Hibiscus tiliaceus - speciation of a pantropical plant with sea-drifted seeds. Mol Ecol. 15(10):2871–2881.
- Tillich M, Lehwark P, Pellizzer T, Ulbricht-Jones ES, Fischer A, Bock R, Greiner S. 2017. GeSeq - versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 45(W1):W6–W11.
- Wick RR, Schultz MB, Zobel J, Jolt KE. 2015. Bandage: interactive visualization of de novo genome assemblies. Bioinformatics. 31(20):3350–3352.
- Zhang R, Wang Y-H, Jin J-J, Stull GW, Bruneau A, Cardoso D, De Queiroz LP, Moore MJ, Zhang S-D, Chen S-Y, et al. 2020. Exploration of plastid phylogenomic conflict yields new insights into the deep relationships of leguminosae. Syst Biol. 69(4):613–622.