
Abstract
Hippuris vulgaris is an aquatic perennial herb distributed worldwide. In this research, the complete chloroplast genome of H. vulgaris was sequenced and assembled. Its complete genome size was 152,698 bp in length. The typical quadripartite structure was shown, which contained a large single-copy region (82,940 bp), a small single-copy region (18,262 bp), and a pair of inverted repeat regions (25,748 bp). The CG content of this genome was 37.6%. A total of 114 genes have been identified in the genome, including 80 protein-coding genes, 30 tRNA genes, and 4 rRNA genes. In addition, 18 genes possessed at least one intron. The phylogenetic analysis indicated that H. vulgaris was nested in Plantaginaceae with 100% bootstrap value and was a sister to Digitalis, Plantago, Hemiphragma, Veronica and Veronicastrum.
Hippuris vulgaris L. is an aquatic plant of worldwide distribution mainly occurred in circumboreal regions with creeping rhizomes, single stem and heterophyll (Chen JR et al; Chen JM et al. Citation2013). The plant grows in streams, lakes, paddy fields, river shores and bogs from 40 m to 5000 m above sea level (Chen JR Citation2000; Lu et al. Citation2016). To date, the complete plastid genome of Hippuris has not been identified. Hence, we sequenced, assembled and annotated the chloroplast genome of H. vulgaris, and tried to ascertain the phylogenetic status of this genome.
Fresh leaves of H. vulgaris were collected from Zaduo, Qinghai, China (N32°47′54.59″, E95°8′55.18″). A specimen as the voucher is deposited in the KUN (Herbarium, Kunming Institute of Botany, Chinese Academy of Sciences; deng7287). Hippuris vulgaris was sequenced by the high-throughput sequencing method with the help of Beijing Novartis Bioinformatics Technology Co., Ltd. The paired-end reads obtained after sequencing were assembled using NOVOPlasty v. 3.7.1 (Dierckxsens et al. Citation2017). The assembled sequence was annotated in MPI-MP CHLOROBOX (https://chlorobox.mpimp-golm.mpg.de/index.html) via GeSeq (Tillich et al. Citation2017) with 2 reference genomes (Plantago depressa Willd. and Plantago lagopus L.), and then manually corrected using Geneious v.9.0.2 (Kearse et al. Citation2012). Eventually, the complete chloroplast genome of H. vularis was submitted to Genbank (GenBank accession number is MT942637).
The genome of H. vulgaris is 152,698 bp in length, which is divided into four parts, including a large single-copy (LSC) region of 82,940 bp, a small single-copy (SSC) region of 18,262 bp and two inverted repeat (IR) regions of 25,748 bp. The CG contents of LSC, SSC, IR and whole genome are 35.7%, 30.8%, 43.1% and 37.6%, respectively. There are 114 unique genes annotated, including 80 protein-coding genes, 30 tRNA genes and 4 rRNA genes. Among them, 6 protein-coding genes, 7 tRNA genes and 4 rRNA genes are duplicated in the IR regions. Of the 18 genes containing introns, three of them have two introns (clpP, ycf3 and rps12).
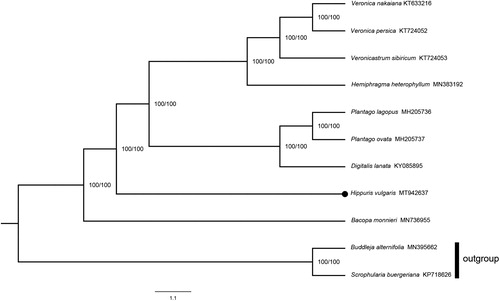
The phylogeny of H. vulgaris was analysis based on the maximum likelihood (ML) method. A total of 11 genomes used for analysis were obtained from NCBI (National Center for Biotechnology Information Search database; https://www.ncbi.nlm.nih.gov/), of which Buddleja alternifolia Maxim. and Scrophularia buergeriana Miq. were selected as outgroups. We aligned the genome with the support of mafft v7.308 (Katoh et al. Citation2002). By using BioEdit v.7.0.5.3 (Hall Citation1999), we checked and corrected the alignment. The nucleotide substitution model was tested by ModelFinder (Kalyaanamoorthy et al. Citation2017). In accordance with the Akaike Information Criterion (AIC), GTR + F + I + G4 was identified as the best appropriate model. The phylogenetic analysis was performed using 1000 replicates in IQ-TREE v.1.6.12 (Trifinopoulos et al. Citation2016). FigTree v.1.4.4 was used to view the phylogenetic tree. The results indicated that H. vulgaris was nested in Plantaginaceae with very strong support (100% bootstrap value). Furthermore, this species was a sister to Digitalis, Plantago, Hemiphragma, Veronica and Veronicastrum ().
Figure 1. The ML phylogenetic tree based on 11 whole genomes. The numbers at the notes are SH-aLRT support (%)/ultrafast bootstrap support (%) from 1000 replicates. The black dot indicates Hippuris vulgaris.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
The data that support the findings of this study are openly available in GenBank at https://www.ncbi.nlm.nih.gov/, reference number MT942637, and in Sequence Read Archive at https://trace.ncbi.nlm.nih.gov/, reference number SRX9471852.
Additional information
Funding
References
- Chen JM, Du ZY, Sun SS, Gituru RW, Wang QF. 2013. Chloroplast DNA phylogeography reveals repeated range expansion in a widespread aquatic herb Hippuris vulgaris in the Qinghai-Tibetan plateau and adjacent areas. PLOS One. 8(4):e60948.
- Chen JR. 2000. Hippuridaceae. In: Ling Y, Shih C, editors. Flora Reipublicae Popularis Sinicae, vol. 53. Beijing: Science Press; p. 144–145.
- Dierckxsens N, Mardulyn P, Smits G. 2017. NOVOPlasty: de novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 45(4):e18.
- Hall TA. 1999. BioEdit: a user-friendly biological sequence alignment program for Windows 95/98/NT. Nucleic Acids Symp Ser. 41:95–98.
- Kalyaanamoorthy S, Minh BQ, Wong TKF, von Haeseler A, Jermiin LS. 2017. ModelFinder: fast model selection for accurate phylogenetic estimates. Nat Methods. 14(6):587–589.
- Katoh K, Misawa K, Kuma K, Miyata T. 2002. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 30(14):3059–3066..
- Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, et al. 2012. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 28(12):1647–1649.
- Lu QX, Zhu JN, Yu D, Xu XW. 2016. Genetic and geographical structure of boreal plants in their southern range: phylogeography of Hippuris vulgaris in China. BMC Evol Biol. 16:34.
- Tillich M, Lehwark P, Pellizzer T, Ulbricht-Jones ES, Fischer A, Bock R, Greiner S. 2017. GeSeq – versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 45(W1):W6–W11.
- Trifinopoulos J, Nguyen LT, Haeseler AV, Minh BQ. 2016. W-IQ-TREE: a fast online phylogenetic tool for maximum likelihood analysis. Nucleic Acids Res. 44(W1):W232–W235.