
Abstract
In this study, we sequenced and annotated the complete mitochondrial genome of Sogatella kolophon (Kirkaldy) (Hemiptera: Delphacidae). The mitogenome was 16,708 bp in length, containing 13 protein-coding genes (PCGs), 22 tRNA genes, two rRNA genes and a control region. All PCGs started with ATN, except for atp6 and nad5, which used noncanonical codon GTG, respectively. All tRNAs could fold into typical clover-leaf secondary structures, with the exception of two trnS genes, lacking the dihydrouridine (DHU) stem.
Keywords:
The delphacid genus Sogatella (Hemiptera: Delphacidae) is a cosmopolitan insect group with some serious pests of rice in Asia and south and central America by direct feeding on the phloem tissues and as vectors of plant virus diseases (Asche and Wilson Citation1990). As one of the most widely distributed species, Sogatella kolophon (Kirkaldy 1907) is the vector of Digitaria striate virus (DSV), Maize sterile stunt virus (MSSV), and Brazilian wheat spike virus (BWSV) (Wilson Citation2005). Here, we report the complete mitochondrial DNA genome of S. kolophon to facilitate better understanding its evolution within Sogatella.
Adults of S. kolophon were collected in Shimen County (N 29.94° and E 110.78°), Hunan Province, China. Voucher specimen (F4-037) and its DNA were deposited in the Institute of Zoology, Chinese Academy of Sciences, Beijing, China. Genomic DNA was extracted using the DNeasy Blood & Tissue Kit (Qiagen, Hilden, Germany) following the manufacturer’s protocols. PCR primers for amplification of the mitogenome were modified from those used in Yu and Liang (Citation2018). Purified PCR products or multiple clones were sequenced directly on an ABI 3730XL DNA Analyzer using BigDye v3.1 (Applied Biosystems, Waltham, MA). After being assembled, the mitogenome sequence was annotated with MitoZ v2.4-alpha (Meng et al. Citation2019), and the annotation of tRNAs was verified by MITOS Web Server (Bernt et al. Citation2013).
The complete mitochondrial genome of Sogatella kolophon (GenBank accession number: MW009064) was 16,708 bp in length. The mitogenome encodes the whole set of genes, including 13 protein-coding genes (PCGs), 22 tRNA genes, two rRNA genes, and a control region, as observed in most insects. Gene rearrangement was detected in the mitogenome of S. kolophon, congruent with those of S. furcifera and S. vibix, in which two gene clusters trnW-trnC-trnY and trnT-trnP-nad6 undergo conversion to trnC-trnW-trnY and nad6-trnP-trnT, respectively (Zhang et al. Citation2014; Huang and Qin Citation2018).
Eleven of all PCGs initiated with the canonical start codons ATG or ATT, except for atp6 and nad5, beginning with GTG, respectively. For 13 PCGs, three stop codons were used: T (atp6, cox1, cox3, nad5), TAG (cox2, cytb, nad1, nad3), and TAA (other five PCGs). All 22 typical tRNA genes were found in the S. kolophon mitogenome, ranging from 56 to 71 bp. The predicted secondary structures of all tRNA genes were typical cloverleaf with exceptions of trnS1 (AGN) and trnS2 (UCN), of which both lack the dihydrouridine (DHU) stem.
The nad4l-nad4 overlap was 7 bp (ATGTTAA) in size, and not identical to that of atp8-atp6 (GTGTTAA). There were a total of nine intergenic spacers throughout the mitogenome of S. kolophon, ranging from 1 bp to 57 bp. The intergenic spacer between trnS2 (UCN) and nad1 was 17 bp in length. As the largest noncoding region, the control region spanning 2304 bp is located between rrnS and trnI. In the control region of S. kolophon, tandem repeat unit was detected, identical with the 21 bp repeat unit in that of the white-backed planthopper Sogatella furcifera (Zhang et al. Citation2014).
Each PCG was translated into amino acid sequence, and aligned with MAFFT v7.394 (Katoh and Standley Citation2013). The concatenated dataset of 13 PCGs was generated via FASconCAT-G v1.04 (Kück and Longo Citation2014). A maximum-likelihood tree () was reconstructed employing the site heterogeneous model PMSF (Wang et al. Citation2018) with 1000 replicates of ultrafast bootstrap in the IQ-TREE v1.6.10 (Nguyen et al. Citation2015). We used the meadow spittlebug Philaenus spumarius (Hemiptera: Aphrophoridae) and Pentastiridius sp. (Hemiptera: Cixiidae) as outgroup.
Figure 1. Maximum-likelihood phylogenetic tree inferred from 13 protein-coding genes. Bootstrap support values are given on nodes. Each family is indicated by vertical lines, and two species from Aphrophoridae (Hemiptera: Cercopoidea) and Cixiidae (Hemiptera: Fulgoroidea) are chosen as outgroup. Sogatella kolophon is marked with asterisk.
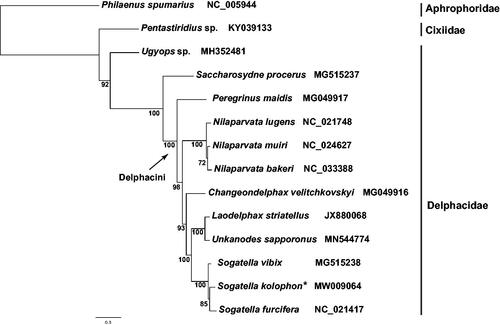
In the clade of Delphacinae, Saccharosydne procerus, representative of Saccharosydnini was sister group to the included species of Delphacini. Nilaparvata lugens was sister to N. muiri+N. bakeri. Unkanodes sapporonus and Laodelphax striatellus were clustered together, indicating their relatively closed relationships. In the genus Sogatella, S. vibix appeared as sister group to S. kolophon and S. furcifera with strong support.
Acknowledgements
The work on which this paper is based was supported by the grant from the National Natural Science Foundation of China (nos. 31970442 and 31961143002, to Z.S.S.).
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
The data that support the findings of this study are openly available in figshare at http://10.6084/m9.figshare.12250331.
References
- Asche M, Wilson MR. 1990. The delphacid genus Sogatella and related groups: a revision with special reference to rice-associated species (Homoptera: Fulgoroidea). Syst Entomol. 15(1):1–42.
- Bernt M, Donath A, Jühling F, Externbrink F, Florentz C, Fritzsch G, Pütz J, Middendorf M, Stadler PF. 2013. MITOS: improved de novo metazoan mitochondrial genome annotation. Mol Phylogenet Evol. 69(2):313–319.
- Huang YX, Qin DZ. 2018. First mitogenome for the tribe Saccharosydnini (Hemiptera: Delphacidae: Delphacinae) and the phylogeny of three predominant rice planthoppers. Eur J Entomol. 115:242–248.
- Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 30(4):772–780.
- Kück P, Longo GC. 2014. FASconCAT-G: extensive functions for multiple sequence alignment preparations concerning phylogenetic studies. Front Zool. 11(1):81.
- Meng G, Li Y, Yang C, Liu S. 2019. MitoZ: a toolkit for animal mitochondrial genome assembly, annotation and visualization. Nucleic Acids Res. 47(11):e63.
- Nguyen LT, Schmidt HA, von Haeseler A, Minh BQ. 2015. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. 32(1):268–274.
- Wang HC, Minh BQ, Susko S, Roger AJ. 2018. Modeling site heterogeneity with posterior mean site frequency profiles accelerates accurate phylogenomic estimation. Syst Biol. 67(2):216–235.
- Wilson SW. 2005. Keys to the families of Fulgoromorpha with emphasis on planthoppers of potential economic importance in the southeastern United States (Hemiptera: Auchenorrhyncha). Fla Entomol. 88(4):464–481.2.0.CO;2]
- Yu F, Liang AP. 2018. The complete mitochondrial genome of Ugyops sp. (Hemiptera: Delphacidae). J Insect Sci. 18:1–13.
- Zhang KJ, Zhu WC, Rong X, Liu J, Ding XL, Hong XY. 2014. The complete mitochondrial genome sequence of Sogatella furcifera (Horváth) and a comparative mitogenomic analysis of three predominant rice planthoppers. Gene. 533(1):100–109.