
Abstract
Sloanea sinensis (Hance) Hu is a tree species and member of the Elaeocarpaceae family. It’s an excellent commercial tree species which has a relatively high net growth as forests. Here, we report the complete chloroplast genome sequence of a Sloanea genus for the first time. The complete chloroplast sequence of S. sinensis is 158,001 bp in length, including a large single copy region (LSC: 88,481 bp) and a small single copy region (SSC: 17,481 bp), the latter of which is separated by a pair of inverted repeat regions (IRs: 26,051 bp). Phylogenetic analysis indicates that the Elaeocarpaceae is a family within the Oxalidales may be more appropriate than belongs to Malvales as traditional plant taxonomy.
Sloanea sinensis (Hance) Hu is an evergreen tree species that belongs to the Sloanea genus, one of two genera that make up the Elaeocarpaceae. S. sinensis is widely distributed in southeastern China and part of southeast Asia, often growing in valleys or near streams. It is an excellent commercial tree species, providing tough, hard and heavy wood suitable for construction, furniture manufacturing etc. Until now, the only DNA sequence data that has been gathered on Sloanea is an incomplete chloroplast sequence from Sloanea australis (Foster et al. Citation2017). Complete sequencing of the chloroplast genome of S. sinensis may lead to a more thorough understanding of Sloanea photosynthesis and resulting wood production.
To obtain S. sinensis total chloroplast DNA, we collected leaf samples from the Yangkou Forest Farm, in Fujian province, China, located at 117.3 O–l18.14E, 26.39–27.12 N (voucher C2020HHX, deposited at Key Laboratory of Forest Genetics & Biotechnology of Ministry of Education of China Nanjing Forestry University). We extracted total DNA and used a HiSeq Xten platform with the PE150 strategy, run by Novogene (Nanjing), for sequencing and generated ∼5.61 G of raw reads. After filtering using SAMtools (Li Citation2011) and Fastp (Chen et al. Citation2018), we obtained ∼5.58 G of clean read data. We used the software package velvet (1.2.10) (Zerbino et al. Citation2009) to assemble the chloroplast genome. We then downloaded reference sequences from NCBI (GenBank accession: NC_044468, NC_039569, NC_037242) and used Geneious Prime (v2020.2.4) and tRNA-SCAN (Chan and Lowe Citation2019) to manually annotate the genomic sequences. Finally, we uploaded the complete S. sinensis chloroplast genome sequence, including annotation, to GenBank filed under accession number MW190090.
Analysis of the complete S. sinensis chloroplast genome sequence, which we found to be 158,001 bp in length, revealed a typical quadripartite construction: a large single copy region (LSC) of 88,481 bp in length, a small single copy region (SSC) of 17,481 bp in length and two inverted repeat regions (IRs), each 26,051 bp in length. The total GC content is ∼37.2%, while the GC content of the LSC, SSC and IRs regions is ∼42.8%, ∼31.5% and ∼35.0%, respectively. We annotated 101 unique genes, including 68 protein-coding genes, 29 tRNA genes and 4 rRNA genes. Because a small number of sequences contained genic mutations leading to premature STOP codons that would disrupt the open reading frame (ORF), we annotated these to be incomplete or pseudogenes. Therefore, the number of functional genes in the S. sinensis chloroplast genome appears to be smaller than in species to which it is directly related.
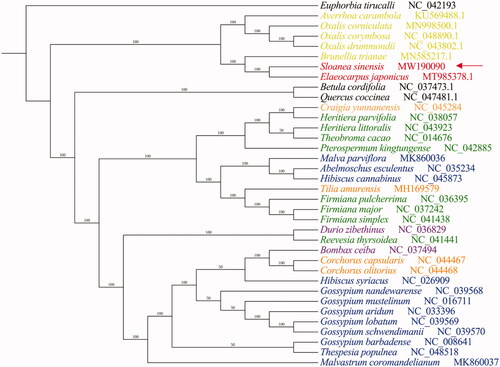
To better understand the phylogenetic relationship of S. sinensis to these direct relatives, we generated a multiple chloroplast genome sequence alignment using Clustal Omega (Sievers and Higgins Citation2018) and from this constructed a phylogenetic tree using MrBayes (default parameters, 50,000 generations) (Ronquist et al. Citation2012). For our analysis, we used 36 plant species, including S. sinensis itself and 35 additional plants from the Malvales order and Oxalidales order, as well as Euphorbia tirucalli, a member of the Euphorbiales order, as an outgroup. Betula cordifolia and Quercus coccinea which belongs to Fagales order to help to distinguish the boundaries. S. sinensis is closely related to E. japonicus. There is no doubt that they belong to Elaeocarpaceae family. Interestingly, base on traditional plant taxonomy which refers to the classification relationship obtained only by plant morphology without the help of molecular genetic information, Elaeocarpaceae family belongs to Malvales, and it belongs to Oxalidales in the taxonomy in NCBI. We found that the four Malvales families do not clearly crystallize out as expected, based on the alignment of their chloroplast genomes (). The result of phylogenetic analysis may more support that S. sinensis is a member of Elaeocarpaceae family which belongs to Oxalidales order. To determine its evolutionary relationships more definitively, the chloroplasts genomes of more Elaeocarpaceae family members require sequencing.
Figure 1. Bayesian inference (BI) generated from a complete chloroplast genome sequence alignment of 37 plant specie, using Euphorbia tirucalli as an outgroup. The number near the clade is posterior probability in percentage format. The Malvales order is subdivided in several plant families, indicated by color coding: the Malvaceae (blue), Tiliaceae (orange), Bombacaceae (purple), Sterculiaceae (green) and Elaeocarpaceae (red), as well as Oxalidales order (yellow).
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
The genome sequence data that support the findings of this study are openly available in GenBank of NCBI at (https://www.ncbi.nlm.nih.gov/) under the accession no. MW190090. The associated BioProject, SRA, and Bio-Sample numbers are PRJNA685222, SRR13254435, and SAMN17080382, respectively.
Additional information
Funding
References
- Chan PP, Lowe TM. 2019. tRNAscan-SE: searching for tRNA genes in genomic sequences. Methods Mol Biol. 1962:1–14.
- Chen S, Zhou Y, Chen Y, Gu J. 2018. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics. 34(17):i884–i890.
- Foster CSP, Sauquet H, van der Merwe M, McPherson H, Rossetto M, Ho SYW. 2017. Evaluating the impact of genomic data and priors on Bayesian estimates of the angiosperm evolutionary timescale. Syst Biol. 66(3):338–351.
- Li H. 2011. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics. 27(21):2987–2993.
- Ronquist F, Teslenko M, van der Mark P, Ayres DL, Darling A, Höhna S, Larget B, Liu L, Suchard MA, Huelsenbeck JP. 2012. MRBAYES 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol. 61:539–542.
- Sievers F, Higgins DG. 2018. Clustal Omega for making accurate alignments of many protein sequences. Protein Sci. 27:135–145.
- Zerbino DR, McEwen GK, Margulies EH, Birney E. 2009. Pebble and rock band: heuristic resolution of repeats and scaffolding in the velvet short-read de novo assembler. PLOS One. 4(12):e8407.