
Abstract
In this study, we present the complete mitogenome and a phylogenetic analysis of Muraenesox cinereus determined by long PCR and primer walking methods. The complete mitochondrial genome is a circular molecule of 17,987 bp in length and contains the same set of 37 mitochondrial genes [13 protein-coding genes, 2 ribosomal RNA (rRNA), and 22 transfer RNA (tRNA)], and a control region as other bony fishes. The base composition of the entire mitogenome showed a slight excess of AT bias. The entire mitogenome data produced in this study provides the genomic resources available for future evolutionary studies.
Muraenesox cinereus belongs to the order Anguilliformes, and is one of the important economic fish species in China. It is mainly distributed along the coast waters of China, Korean Peninsula, and Japan, and also the east coast of Africa in the Indian Ocean (Ni and Wu Citation2006). It is a fierce fish live in inshore warm bottom water and usually inhabited in the seawater with sediment or rock reef bottom. No relevant research on M. cinereus has been documented till now. Therefore, it is highly important to obtain the complete mitochondrial genome of M. cinereus for further studies.
In this study, one M. cinereus specimen (NO.HM20040901) collected from the Coastal survey of Jiangsu Province (121°45′E, 32°15′N) on April 2020 and stored in the herbarium of Jiangsu Marine Fisheries Research Institute.
The complete mitogenome of M. cinereus is sequenced to be 17,987 bp in length (GenBank accession number: MT855985) and shares most similarity on gene content and structure with most vertebrates (Boore Citation1999; Wolstenholme Citation1992), but the ND6 change places with Cyt b. It consists of 13 typical vertebrate protein-coding genes, 22 tRNA genes, 2 rRNA genes (12S rRNA and 16S rRNA), and 2 major noncoding regions (control region and L-strand replication origin). Most mitochondrial genes of M. cinereus are encoded on the H-strand, except for ND6 and eight tRNA (Gln, Ala, Asn, Cys, Tyr, Ser-UCN, Glu, and Pro) genes that are encoded on the L-strand. The overall nucleotide composition of the heavy strand is 27.87% T, 23.54% C, 31.12% A, and 17.47% G, with anti-G bias and a slight excess of AT as the observation in most fishes (Miya et al. Citation2001, Citation2003). The 13 protein-coding genes are totally 11,487 bp in size, accounting for 63.86% of the whole mitogenome. The M. cinereus mitogenome also contains a small subunit rRNA (12S rRNA) and a large subunit rRNA (16S rRNA), which are 962 and 1699 bp in length, respectively.
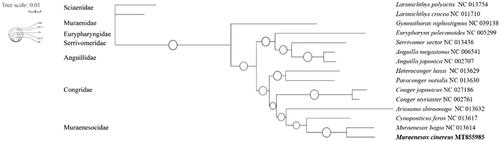
To investigate the phylogenetic relationships of M. cinereus and other 11 species from Anguilliformes, phylogenetic trees are obtained using Maximum-Likelihood (ML) analyses based on entire COI sequences. Three species in the order Saccopharyngiformes and Perciformes are used as outgroup (). The topology of phylogeny trees for 12 Anguilliformes species shows that family Muraenesocidae is a single group. Unexpectedly, the Eurypharynx pelecanoides in order Saccopharyngiformes separate the order Anguilliformes into two parts. Nowdays there are not enough sequences of the family Muraenesocidae can be used for analysis, therefore more details of the topology of phylogeny trees need to be studied and further investigation will be needed to reveal the reason for this unexpected phenomenon.
Figure 1. Phylogenetic tree using the COI sequences. The sequences are downloaded from GenBank and the phylogenic tree is constructed by maximum-likelihood (ML) method. The posteriori probability was presented as the proportion of white circles. The genomic sequences obtained in this study are shown by bold type.
Acknowledgments
The authors would like to thank Wenming Yang and Junhui Li for their helpful suggestions.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
The data that support the findings of this study are openly available in GenBank of NCBI at https://www.ncbi.nlm.nih.gov, reference number MT855985.
Additional information
Funding
References
- Boore JL. 1999. Animal mitochondrial genomes. Nucleic Acids Res. 27(8):1767–1780.
- Miya M, Kawaguchi A, Nishida M. 2001. Mitogenetic exploration of higher teleostean phylogenies: a case study for moderate-scale evolutionary genomics with 38 newly determined complete mitochondrial DNA sequences. Mol Biol Evol. 18(11):1993–2009.
- Miya M, Takeshima H, Endo H, Ishiguro NB, Inoue JG, Mukai T, Satoh TP, Yamaguchi M, Kawaguchi A, Mabuchi K, et al. 2003. Major patterns of higher teleostean phylogenies: a new perspective based on 100 complete mitochondrial DNA sequences. Mol Phylogenet Evol. 26(1):121–138.
- Ni Y, Wu H. 2006. Fishes of Jiangsu province. Beijing, China: China Agriculture Press.
- Wolstenholme DR. 1992. Animal mitochondrial DNA: structure and evolution. Int Rev Cytol. 141:173–216.