
Abstract
We have determined the second mitochondrial genome of Myotis bombinus Thomas, 1906 in mainland of Korea. The circular mitogenome of M. bombinus is 17,035 bp long which is slightly shorter than that of the previous mitogenome of M. bombinus. It includes 13 protein-coding genes (PCGs), two ribosomal RNA genes, and 22 transfer RNAs. The base composition was AT-biased (66.1%). Fifty single nucleotide polymorphisms and 14 insertions were identified between two mitogenomes of M. bombinus. Phylogenetic trees show that both M. bominus mitogenomes are clustered in one clade.
Myotis bombinus Thomas, 1906 was distributed in Russia, China, Mongolia, Japan, and Korea (Jargalsaikhan Citation2016). Especially, M. bombinus inhabits whole Korean peninsula including Jejudo island (Won and Smith Citation1999; Park et al. Citation2015; Jo et al. Citation2018). Most typical habitats of M. bombinus are forest, cave, and subterranean habitats (Funakoshi Citation1991; Kim S-S et al. Citation2014); however, M. bombinus became near threatened (NT) species in the world because of loss of these habitats world-widely (Fukui et al. Citation2019), requiring intensive researches of this species in various ways.
We completed the mitogenome of M. bombinus, collected at the Jeombong Mountain, located in Jindong-myeon, Inje-gun, Gangwon-do, Republic of Korea (38°2′32.12′′N, 128°28′23.77′′E). DNA was extracted using DNeasy Blood & Tissue Kit (QIAGEN, Hilden, Germany). Raw sequences from Illumina NovaSeq6000 (Macrogen Inc., Seoul, Korea) were filtered by Trimmomatic v0.33 (Bolger et al. Citation2014) and de novo assembled by Velvet v1.2.10 (Zerbino and Birney Citation2008), SOAPGapCloser v1.12 (Zhao et al. Citation2011), BWA v0.7.17 (Li Citation2013), and SAMtools v1.9 (Li et al. Citation2009) under the environment of Genome Information System (GeIS; http://geis.infoboss.co.kr/; Park et al., in preparation). Geneious R11 v11.1.5 (Biomatters Ltd, Auckland, New Zealand) was used to annotate our mitogenome based on M. bombinus mitogenome (NC_029342; Kim Y-K et al. Citation2017). DNA sample and tissues of M. bombinus were deposited in InfoBoss Cyber Herbarium (IN; Kim S-S; Voucher numbers are IBS-00019 and IB-40004).
M. bombinus mitogenome (GenBank accession is MT985383) is 17,035 bp, slightly shorter than that of the previous sequenced mitogenome of M. bombinus (NC_029342; 17,128 bp; Kim Y-K et al. Citation2017). It contains 13 protein-coding genes (PCGs), 22 transfer RNAs, and two ribosomal RNAs. Its AT ratio is 64.7%.
Based on pair-wise alignment of two M. bombinus mitogenomes, 50 single nucleotide polymorphisms (SNPs) and 14 insertions of which length was 95 bp in total were found against M. bombinus mitogenome isolated in Jejudo island (NC_029342). Twenty out of the 50 SNPs were located in PCGs, displaying that three were non-synonymous SNPs in ND2 and COX1 and 17 out of 50 SNPs were synonymous SNPs in eight PCGs. Number of polymorphic sites of four Myotis petax mitogenomes (Hwang et al. Citation2016) is 51, which is almost similar to number of SNPs of M. bombinus. In addition, the four samples of M. petax are also isolated from Korean peninsula (Hwang et al. Citation2016), indicating that genetic diversity of M. bombinus mitogenomes in Korea is similar to that of M. petax.
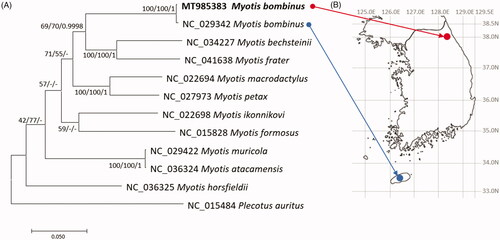
We inferred phylogenetic relationship of eleven Myotis and one Plecotus mitogenomes, based on trimmed alignment of mitogenome sequences by MAFFT v7.450 (Katoh and Standley Citation2013). Bootstrapped neighbor-joining, maximum-likelihood, and Bayesian Inference phylogenetic trees with MEGA X (Kumar et al. Citation2018) and MrBayes v3.2.7a (Huelsenbeck and Ronquist Citation2001), respectively, were constructed. Phylogenetic trees display that two M. bombinus mitogenomes are clustered in one clade (). Taken together, genetic diversity of M. bombinus in Korean peninsula can be estimated, especially for genetic difference between Korean peninsula and Jejudo island.
Figure 1. (A) Maximum-likelihood (1,000 bootstrap repeats), neighbor-joining (10,000 bootstrap repeats), and Bayesian Inference (1,100,000 generations) phylogenetic trees of 11 Myotis mitochondrial genomes and one Plecotus mitogenomes: two M. bombinus, M. bechsteinii, M. frater, M. macrodactylus, M. petax, M. ikonnikovi, M. horsfieldii, M. muricola, M. atacamensis, M. formosus, and P. auratus. Phylogenetic tree was drawn based on maximum-likelihood tree. The numbers above branches indicate bootstrap support values of maximum-likelihood and neighbor-joining phylogenetic trees and posterior probability value of Bayesian Inference tree, respectively. (B) displays geographical location of two M. bombinus samples: red circle indicates the sample used in this study and blue circle means the sample used in the previous study (NC_029342; Kim et al. Citation2017).
Disclosure statement
The authors declare that they have no competing interests.
Data availability statement
Mitochondrial genome sequence can be accessed via accession number MT985383 in NCBI GenBank.
Additional information
Funding
References
- Bolger AM, Lohse M, Usadel B. 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 30(15):2114–2120.
- Fukui D, Sano A, Kruskop SV. 2019. Myotis bombinus. The IUCN Red List of threatened species. [accessed 2020 Sept 11]. https://www.iucnredlist.org/species/14149/22061650.
- Funakoshi K. 1991. Reproductive ecology and social dynamics in nursery colonies of the Natterer’s bat Myotis nattereri bombinus. J Mammal Soc Jpn. 15(2):61–71.
- Huelsenbeck JP, Ronquist F. 2001. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics. 17(8):754–755.
- Hwang JY, Jin GD, Park J, Lee SG, Kim EB. 2016. Complete sequences of eastern water bat, Myotis petax (Chiroptera; Microchiroptera; Vespertilionidae) mitogenome. Mitochondrial DNA Part A. 27(5):3715–3716.
- Jargalsaikhan A. 2016. Bat study in the Kharaa region, Mongolia. J Asia Pac Biodivers. 9(2):107–115.
- Jo YS, Baccus JT, Koprowski JL. 2018. Mammals of Korea: a review of their taxonomy, distribution and conservation status. Zootaxa. 4522(1):1–216.
- Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 30(4):772–780.
- Kim S-S, Choi Y-S, Yoo J-C, Ecosystem Service Research Team, National Institute of Ecology, Seocheon 325-813, Korea. 2014. The thermal preference and the selection of hibernacula in seven cave-dwelling bats. Korean J Ecol Environ. 47(4):258–272.
- Kim Y-K, Park S-G, Kim T-W, Park J-H, Adhikari P, Kim G, Park S-M, Lee J-W, Oh D-J, Han S-H, et al. 2017. Complete mitochondrial genome of the far eastern Myotis, Myotis bombinus (Chiroptera, Vespertilionidae). Mitochondrial DNA Part A. 28(2):267–268.
- Kumar S, Stecher G, Li M, Knyaz C, Tamura K. 2018. MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol. 35(6):1547–1549.
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R. 2009. The sequence alignment/map format and SAMtools. Bioinformatics. 25(16):2078–2079.
- Li H. 2013. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv preprint arXiv:13033997.
- Park SG, Kim YK, Kim TW, Park JH, Adhikari P, Kim GR, Park SM, Lee JW, Han SH, Oh HS. 2015. A study on the distribution of bats (Chiroptera) in Jeju Island, Korea. Korean J Environ Biol. 33(4):394–402.
- Won C, Smith KG. 1999. History and current status of mammals of the Korean peninsula. Mammal Rev. 29(1):3–33.
- Zerbino DR, Birney E. 2008. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 18(5):821–829.
- Zhao QY, Wang Y, Kong YM, Luo D, Li X, Hao P. 2011. Optimizing de novo transcriptome assembly from short-read RNA-Seq data: a comparative study. BMC Bioinf. 12(Suppl 14):S2.