
Abstract
Scapania ampliata Steph. is the endemic species in East Asia. To investigate intraspecific variations on mitochondrial genomes of S. ampliata, we completed mitochondrial genome of S. ampliata isolated in Korea. It is 143,664 bp long and contains 73 genes (41 protein-coding genes, three rRNAs, 28 tRNAs, and one pseudogene). 823 single nucleotide polymorphisms (SNPs; 0.057%) and 2,242 insertions and deletions were identified between two S. ampliata mitochondrial genomes, which is large number of intraspecific variations in comparison to the other cases of Bryophyte mitochondrial genomes. Phylogenetic trees show that S. ampliata is clustered with those of two Scapania species with high supportive values.
Scapania ampliata Steph. was described by Stephani based on a specimen collected by U. Faurie from the Honshu in Japan (Stephani Citation1897). S. ampliata is the endemic species in East Asia, distributed in Korean Peninsula (North and South), Japan, and China (Choi et al. Citation2012). Scapania ampliata grows on humus, shaded rocks, or decaying wood, and commonly mixture Dounia plicata (Lindb.) Konstant. et Vilent, Bazzania denudata (Torr. Ex Lindb.) Trevis., and Lepidozia reptans (L.) Dumort. in the subalpine region. Moreover, it is endemic to East Asia, which can be used for understanding its genetic features as well as Scapaniaceae in the world. Here, we completed the mitogenome of S. ampliata for understanding its intraspecific variations and phylogenetic features together with its chloroplast genome already sequenced (Choi, Kwon, et al. Citation2020).
The plants of S. ampliata collected in Taebaek city, Korea (Voucher in Jeonbuk National University Herbarium (JNU); Contact: Seung Se Choi; [email protected]; S.S. Choi, CS-1910671a; 37.101486N, 128.917547E) was used for extracting DNA with DNeasy Plant Mini Kit (QIAGEN, Hilden, Germany). Genome sequencing was performed using NovaSeq6000 at Macrogen Inc., Korea. Mitochondrial genome was completed by Velvet v1.2.10 (Zerbino and Birney Citation2008), SOAPGapCloser v1.12 (Zhao et al. Citation2011), BWA v0.7.17 (Li Citation2013), and SAMtools v1.9 (Li et al. Citation2009) under the environment of Genome Information System (GeIS; http://geis.infoboss.co.kr/; Park et al., in preparation). We manually confirmed all bases of the assembled mitochondrial genome were correct using tview mode of SAMtools v1.9 (Li et al. Citation2009). The average depth of this assembled mitochondrial genome displays 182.23x. Geneious R11 v11.0.5 (Biomatters Ltd, Auckland, New Zealand) was used for annotation based on Scapania griffithii mitochondrial genome (MK230961; Dong et al. Citation2019).
The mitochondrial genome of S. ampliata (GenBank accession is MT755612) is 143,664 bp long, which is longer than that of Scapania ornithopodioides (142,992 bp; MK230950; Dong et al. Citation2019) by 672 bp. It contains 72 genes (42 protein-coding genes, three rRNAs, and 27 tRNAs) and overall GC content is 45.0%. Gene order of S. ampliata mitogenome is the same as that of S. griffithii. 823 single nucleotide polymorphisms (SNPs; 0.057%) and 2,242 insertions and deletions (INDELs; 1.56%) were identified from the two Scapania mitogenomes. These interspecific variations are much higher than those of Marchandia polymorpha subsp. ruderalis (7 SNPs; 0.0038%; Kwon et al. Citation2019b), Riccia fluitans (18 SNPs; 0.0097% and 19 INDELs; 0.010%; Min et al. Citation2020), Dumortiera hirsuta (12 SNPs; 0.0067% and 24 INDELs; 0.013% with one big inversion; Dong et al. Citation2019; Kwon et al. Citation2019a), and Monosolenium tenerum (14 SNPs; 0.0075% and 7 INDELs; 0.0037%; Dong et al. Citation2019) as expected, which is also congruent to those of angiosperm species, such as Liriodendron tulifipera (365 SNPs; 0.066%; 2,117 INDELs; 0.38%; Park et al. Citation2019), Arabidopsis thaliana (64 SNPs; 0.017% and 1,089 INDELs; 0.30%; Park et al., in preparation), and Rosa rugosa (124 SNPs; 0.041%, 769 INDELs; 0.25%; Park et al. Citation2020). However, those are similar to those of Wiesnerella denudata (149 SNPs; 0.080% and 3,033 INDELs; 1.62%; Choi, Min, et al. Citation2020). It presents that numbers of intraspecific or inter-specific variations are various along with Bryophyte species.
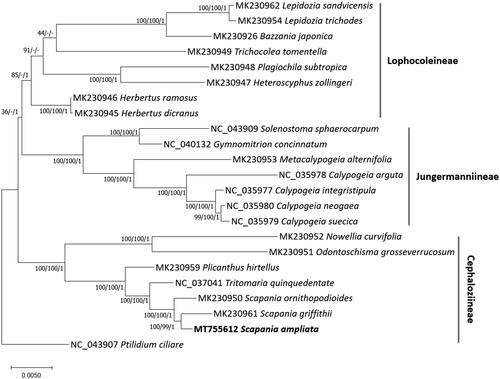
Twenty-three complete mitochondrial genomes including S. ampliata mitogenome and one outgroup species, Ptilidium ciliare, were used for drawing Neighbor-Joining (bootstrap repeat is 10,000), Maximum-Likelihood (bootstrap repeat is 1,000), and Bayesian Inference phylogenic trees (Number of generations is 1,100,000) with MEGA X (Kumar et al. Citation2018) and MrBayes v3.2.7a (Huelsenbeck and Ronquist Citation2001), respectively, based on alignments of 38 conserved protein-coding genes based on the annotation of the used mitochondrial genomes by MAFFT v7.450 (Katoh and Standley Citation2013). Phylogenetic trees present that three Scapania mitogenomes were clustered with high supportive values of the three trees (). In addition, topologies in the Lophocoleineae suborder clade display disconcordance among three phylogenetic trees (). Three suborders, Lophocoleineae, Jungermanniineae, and Cephaloziineae, were clustered monophyletically supported by three different phylogenetic trees ().
Figure 1. Neighbor-Joining (bootstrap repeat is 10,000), Maximum-Likelihood (bootstrap repeat is 1,000), and Bayesian Inference (Number of generations is 1,100,000) phylogenetic trees of twenty-three complete mitochondrial genomes. Phylogenetic tree was drawn based on Maximum-lLkelihood phylogenetic tree. The numbers above branches indicate bootstrap support values of Maximum-Likelihood, Neighbor-Joining, and Bayesian Inference phylogenetic trees, respectively.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
Mitochondrial genome sequence can be accessed via accession number MT755612 in GenBank of NCBI at https://www.ncbi.nlm.nih.gov. The associated BioProject, SRA, and Bio-Sample numbers are PRJNA691085, SAMN17277783, and SRR13399294, respectively.
Additional information
Funding
References
- Choi SS, Bakalin VA, Sun B-Y. 2012. Scapania and Macrodiplophyllum in the Russian Far East. Bot Pac. 01(1):31–95.
- Choi SS, Kwon W, Park J. 2020. The complete chloroplast genome of Scapania ampliata Steph., 1897 (Scapaniaceae, Jungermanniales). Mitochondrial DNA B Resour. 5(3):2890–2910.
- Choi SS, Min J, Kwon W, Park J. 2020. The complete mitochondrial genomeof Wiesnerella denudata (Mitt.) Steph.(Wiesnerellaceae, Marchantiophyta): large number of intraspecific variations on mitochondrial genomes of W. denudata. Mitochondrial DNA Part B. 5(3):3371–3669.
- Dong S, Zhao C, Zhang S, Zhang L, Wu H, Liu H, Zhu R, Jia Y, Goffinet B, Liu Y. 2019. Mitochondrial genomes of the early land plant lineage liverworts (Marchantiophyta): conserved genome structure, and ongoing low frequency recombination. BMC Genomics. 20(1):953.
- Huelsenbeck JP, Ronquist F. 2001. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics. 17(8):754–755.
- Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 30(4):772–780.
- Kumar S, Stecher G, Li M, Knyaz C, Tamura K. 2018. MEGA X: Molecular Evolutionary Genetics Analysis across computing platforms. Mol Biol Evol. 35(6):1547–1549.
- Kwon W, Kim Y, Park J. 2019a. The complete mitochondrial genome of Dumortiera hirsuta (Sw.) Nees (Dumortieraceae, Marchantiophyta). Mitochondrial DNA Part B. 4(1):1586–1587.
- Kwon W, Kim Y, Park J. 2019b. The complete mitochondrial genome of Korean Marchantia polymorpha subsp. ruderalis Bischl. & Boisselier: inverted repeats on mitochondrial genome between Korean and Japanese isolates. Mitochondrial DNA Part B. 4(1):769–770.
- Li H. 2013. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv preprint arXiv:13033997.
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R. 2009. The sequence alignment/map format and SAMtools. Bioinformatics. 25(16):2078–2079.
- Min J, Kwon W, Xi H, Park J. 2020. The complete mitochondrial genome of Riccia fluitans L. (Ricciaceae, Marchantiophyta): investigation of intraspecific variations on mitochondrial genomes of R. fluitans. Mitochondrial DNA B Resour. 5(2):1220–1222.
- Park J, Kim Y, Kwon M. 2019. The complete mitochondrial genome of tulip tree, Liriodendron tulipifera L. (Magnoliaceae): intra-species variations on mitochondrial genome. Mitochondrial DNA Part B. 4(1):1308–1309.
- Park J, Xi H, Kim Y, Nam S, Heo K-I. 2020. The complete mitochondrial genome of newspecies candidate of Rosa rugosa (Rosaceae). Mitochondrial DNA Part B. 5(3):3435–3455.
- Stephani F. 1897. Species Hepaticarum, continuation. Bull Herb Boissier. 7(5):381–407.
- Zerbino DR, Birney E. 2008. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 18(5):821–829.
- Zhao Q-Y, Wang Y, Kong Y-M, Luo D, Li X, Hao P. 2011. Optimizing de novo transcriptome assembly from short-read RNA-Seq data: a comparative study. BMC Bioinf. 12(14):S2.