
Abstract
Elymus kamoji (Ohwi) S. L. Chen is a perennial herb, had high grazing value and were important forage resources, the study of E. kamoji chloroplast genome (cp genome) provides an important basis for the study of chloroplast genetic engineering and system evolution. Its chloroplast genome was 135,075 bp in length, containing a pair of inverted repeated (IR) regions (20,813 bp), separated by a large single copy region (LSC) of 80,681 bp, and a small single copy (SSC) region of 12,768 bp. Moreover, a total of 129 functional genes were annotated, including 83 mRNA, 38 tRNA genes, and 8 rRNA genes. The phylogenetic relationships of 15 species indicated that E. kamoji was closely related to Elymus sibiricus. This study might contribute to provide a theoretical basis for species identification and biological research.
Elymus kamoji (Ohwi) S. L. Chen, belonging to the Roegneria (Triticeae), which is a perennial herb mainly distributed in China, Japan and Korea (Sun et al. Citation2017). Because of the excellent genes of disease resistance and stress resistance and high feed value, E. kamoji is considered to be an important natural gene bank for improvement of wheat related plants and forage breeding (Cai Citation2002). However, the evolutionary relationship and systematic status of E. kamoji has always been controversial. With the development of molecular biology, phylogenetic analysis has been used as an effective method to reveal the origin and evolution of species and clarify the genetic relationship between different genera and species (Baum et al. Citation2015). The chloroplast genome (cp genome) contains a large number of functional genes, and their application values in the study of species identification and phyletic evolution have been widely accepted and gradually recognized by researchers (Daniell et al. Citation2016). In this study, the cp genome sequencing of E. kamoji (GenBank accession number: MW043483) was done by Illumina NovaSeq platform, and the cpDNA sequence structure was analyzed, which would provide a basis for the judgment of the relationship of related species and genera, and provides more condition support for plant phylogeny research.
The fresh young leaves of E. kamoji were collected in Xihai Town, Haibei Prefecture, Qinghai Province, China (E100°52.848′, N36°59.36′). The voucher specimen was kept in Herbarium of Low Temperature Bank of Germplasm Resources of Qinghai Academy of Animal Husbandry and Veterinary Sciences (contact person and email: Xiaoxing Wei, [email protected]) under the voucher number 09-110. The total genomic DNA of E. kamoji was extracted from the fresh leaves with a modified CTAB method (Li et al. Citation2018). After a series of processing, the genomic DNA was amplified by PCR was used to generate libraries with an average insert size of 300 bp and the genome sequencing was performed using the Illumina HiSeq Platform (Illumina, San Diego, CA) at Gene pioneer Biotechnologies Inc., Nanjing, China. Finally, about 7.31 GB of clean data were generated and the SPAdes (Bankevich et al. Citation2012) was used to genome assembly, based on the clean data. The assembled genome was annotated using CpGAVAS (Liu et al. Citation2012).
The complete cp genome of E. kamoji was 135,075 bp in length, exhibiting a typical quadripartite structure including a pair of inverted repeated (IRA and IRB) regions (20, 813 bp) that are separated by a large single copy (LSC) region of 80,681 bp, and a small single copy (SSC) region of 12,768 bp. The GC content of the whole cp genome was 38.33%. A total of 129 functional genes were annotated, including 83 protein-coding genes (mRNA), 38 tRNA genes, and 8 rRNA genes. The protein-coding genes, tRNA genes, and rRNA genes account for 64.34%, 29.46% and 6.20% of all annotated genes, respectively.
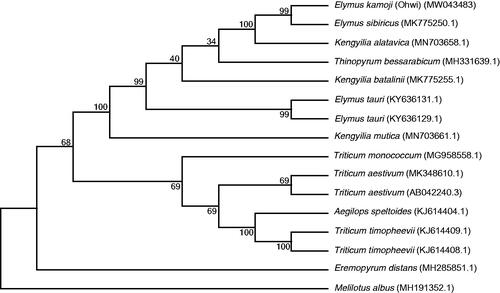
To confirm the phylogenetic position of E. kamoji, the cp genome sequences of E. kamoji and 15 other species were aligned with MAFFT version 7 (https://mafft.cbrc.jp/alignment/software/algorithms/algorithms.html), and the maximum- likelihood (ML) tree was constructed based on the genome sequences of E. kamoji, 14 reported Gramineae species and one outgroup species Leguminosae species (Melilotus albus) using MEGA7.0 (Kumar et al. Citation2016) under the model of Kimura 2-parameter with 1000 bootstrap. The results showed that E. kamoji was closely related to E. sibiricus ().
Figure 1. Phylogenetic relationships of 16 species based on complete chloroplast genome using the Maximum-Likelihood methods. The bootstrap values were based on 1000 replicates and are shown next to the branches.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
The genome sequence data that support the findings of this study are openly available in GenBank of NCBI at (https://www.ncbi.nlm.nih.gov/) under the accession no.MW043483. The associated Bio-Project, SRA, and Bio-Sample numbers are PRJNA680463, SRR13385763, and SAMN16880839 respectively.
Additional information
Funding
References
- Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, Pyshkin AV, et al. 2012. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 19(5):455–477.
- Baum BR, Edwards T, Johnson DA. 2015. Diversity within the Genus Elymus (Poaceae: Triticeae) as Investigated by the Analysis of the Nr5s Rdna variation in species with St and H Haplomes. Mol Genet Genom. 290(1):329–342.
- Cai BL. 2002. Geographical distribution of Roegneria C. Koch (Poaceae). Acta Botanica Boreali-Occidentalia Sinica. 22(4):189–199. Chinese.
- Daniell H, Lin CS, Yu M, Chang WJ. 2016. Chloroplast genomes: diversity, evolution, and applications in genetic engineering. Genome Biol. 17(1):1–29.
- Kumar S, Stecher G, Tamura K. 2016. Mega7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol. 33(7):1870–1874.
- Li X, Li YF, Zang MY, Li MZ, Fang YM. 2018. Complete chloroplast genome sequence and phylogenetic analysis of Quercusacutissima. Int J Mol Sci. 19(8):2443–2459.
- Liu C, Shi L, Zhu Y, Chen H, Zhang J, Lin X, Guan X. 2012. CpGA V AS, an integrated web server for the annotation, visualization, analysis, and GenBank submission of completely sequenced chloroplast genome sequences. BMC Genomics. 13(1):715.
- Sun ZH, Zhang Y, Deng MQ, Jiang ZX, Jiao ZF, Zhou YH, Zhang HQ. 2017. Cytogenetic and Morphological Studies on Seven Populations of RoegneriakamojiOhwi (Triticeae: Poaceae). J Plant Genet Resour. 18(6):1032–1038.