
Abstract
Metarhizium album, with a narrow host range, is an entomopathogenic fungus in the family Clavicipitaceae. Its nuclear genome has been sequenced, whereas its mitogenome is still unknown. In this study, the complete mitogenome of M. album was assembled and annotated. This circular mitogenome was 68,425 bp in length and encodes two rRNA genes, 26 tRNA genes, 14 standard protein-coding genes of the oxidative phosphorylation system, and seven intergenic ORFs. A total of 23 introns invaded ten genes, including atp9 (1 intron), cob (2), cox1 (9), cox2 (1), nad1 (1), nad2 (2), nad3 (1), nad4 (1), nad5 (2), and rnl (3). Except for one group II intron (i.e. mL2060), others were all group I introns and involved four subgroups (i.e. IA, IB, IC2 and ID). Phylogenetic analysis based on mitochondrial nucleotide sequences confirmed M. album in the family Clavicipitaceae, being closely related to its congeneric Metarhizium rileyi.
Metarhizium album Petch is an entomopathogenic fungus within the family Clavicipitaceae (Ascomycota: Hypocreales). It was first described as occurring on a leaf-hopper on rice in Sri Lanka (Petch Citation1931). The fungus has a much more limited host range than its congeneric Metarhizium anisopliae and is specific for hemipteran insects, mostly from the family Cicadellidae (Rombach et al. Citation1987). The nuclear genome of M. album was previously reported (Hu et al. Citation2014). Nevertheless, the mitogenome information of the fungus is still lacking. Herein, we present the complete mitogenome of M. album.
Metarhizium album strain ARSEF 1941 was recovered from Nephotettix virescens (Hemiptera: Cicadellidae) on rice in Roxas, Palawan, Philippines (N10.32, E119.26) and deposited in USDA-ARS Collection of Entomopathogenic Fungal Cultures (https://www.ars.usda.gov/, Melanie Filiatrault, [email protected]). Total DNA was isolated from mycelia and stored at −80 °C at the Laboratory of Microbial Evolutionary Biology at Shanxi University, China. The extracted DNA was fragmented by sonication to a size of ∼280 bp, followed by sequencing on an Illumina Xten platform in 2 × 150 bp reads. Mitogenome was de novo assembled from clean reads using NOVOPlasty (Dierckxsens et al. Citation2017) and then annotated as described previously (Zhang et al. Citation2017). Introns were named according to a newly suggested nomenclature (Zhang and Zhang Citation2019).
The mitogenome of M. album (GenBank accession: MW448543) was a circular molecule of 68,425 bp with AT content of 73.98%. This mitogenome encoded two rRNAs (rnl and rns), 26 tRNAs, 14 conserved proteins of the oxidative phosphorylation system (nad1-6, 4L; cob; cox1-3, and atp6, 8, 9), and seven intergenic ORFs. These tRNA genes coded for all 20 standard amino acids. All intergenic ORFs encoded hypothetical proteins.
Ten genes were invaded by a total of 23 introns, including atp9 (1 intron), cob (2), cox1 (9), cox2 (1), nad1 (1), nad2 (2), nad3 (1), nad4 (1), nad5 (2), and rnl (3). Except for one group II intron (i.e. mL2060), all other introns belonged to the group I intron family but fell into four specific subgroups, namely IA (2 introns), IB (11), IC2 (5), and ID (4). Except for one intron (i.e. nad1P636), all other introns contained putative ORFs encoding for ribosomal protein S3, LAGLIDADG or GIY-YIG homing endonucleases, or hypothetical proteins. Evidence for the degeneration of the ORF-lacking intron (nad1P636) was obvious because of frame shifts and stop codon mutations.
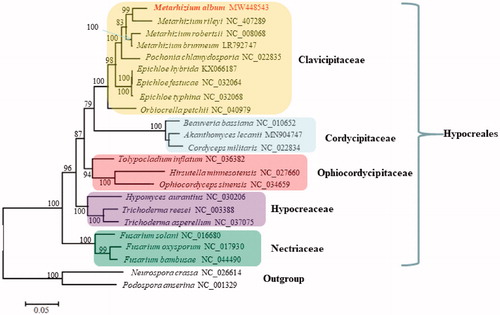
Phylogenetic analysis based on mitochondrial nucleotide sequences by the Maximum Likelihood approach confirmed M. album as a member in Clavicipitaceae (). The fungus was closely related to its congeneric Metarhizium rileyi and their clustering received 99% support value. Actually, the phylogenetic tree deduced herein was largely congruent with the tree inferred from nuclear multi-locus analyses (Sung et al. Citation2008), with all species of the same family clustering as a separate clade ().
Figure 1. Phylogenetic analysis of Hypocreales species based on mitochondrial nucleotide sequences. We used all species of Clavicipitaceae and representative species of all other families with available mitogenomes in Hypocreales in January, 2021. Two Sordariales species (Podospora anserine and Neurospora crassa) were used as outgroups. The whole mitogenome sequences (or exonic sequences in cases with alignment difficulties) of these species were aligned and trimmed using the HomBlocks pipeline (Bi et al. Citation2018), resulting in an alignment of 6911 characters. Phylogenetic reconstruction was performed using the maximum likelihood approach as implemented in RAxML v8.2.12 (Stamatakis Citation2014). Support values were given for nodes that received bootstrap values ≥ 70%. GenBank accession numbers followed after fungal taxon names.
Acknowledgments
We thank the High Performance Simulation Platform of Shanxi University for providing computing resource.
Disclosure statement
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the article.
Data availability statement
The mitochondrial genome sequence data that support the findings of this study are openly available in GenBank of NCBI under the accession no. MW448543 (https://www.ncbi.nlm.nih.gov/nuccore/MW448543).
Additional information
Funding
References
- Bi G, Mao Y, Xing Q, Cao M. 2018. HomBlocks: a multiple-alignment construction pipeline for organelle phylogenomics based on locally collinear block searching. Genomics. 110(1):18–22.
- Dierckxsens N, Mardulyn P, Smits G. 2017. NOVOPlasty: de novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 45(4):e18.
- Hu, Xiao, Xiao, Guohua, Zheng, Peng, Shang, Yanfang, Su, Yao, Zhang, Xinyu, Liu, Xingzhong, Zhan, Shuai, St. Leger, Raymond J., Wang, Chengshu. 2014. Trajectory and genomic determinants of fungal-pathogen speciation and host adaptation. Proc Natl Acad Sci USA. 111(47):16796–16801.
- Petch T. 1931. Notes on entomogenous fungi. Trans Br Mycol Soc. 16(1):55–75.
- Rombach MC, Humber RA, Evans HC. 1987. Metarhizium album, a fungal pathogen of leaf- and planthoppers of rice. Trans Br Mycol Soc. 88(4):451–459.
- Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies.Bioinformatics. 30(9):1312–1313.
- Sung GH, Poinar Jr GO, Spatafora JW. 2008. The oldest fossil evidence of animal parasitism by fungi supports a Cretaceous diversification of fungal-arthropod symbioses. Mol Phylogenet Evol. 49(2):495–502.
- Zhang S, Zhang Y-J. 2019. Proposal of a new nomenclature for introns in protein-coding genes in fungal mitogenomes. IMA Fungus. 10:15.
- Zhang Y-J, Yang X-Q, Zhang S, Humber RA, Xu J. 2017. Genomic analyses reveal low mitochondrial and high nuclear diversity in the cyclosporin-producing fungus Tolypocladium inflatum. Appl Microbiol Biotechnol. 101(23–24):8517–8531.