
Abstract
Flesh-footed shearwater (Ardenna carneipes) is recognized as vulnerable seabird species in Western Australia and New South Wales, Australia, and its genetic variability and a well-resolved phylogeny is imperative for the species’ conservation. Here, we report the first sequenced mitogenome of the Australian A. carneipes. The mitogenome of A. carneipes was 16,370 bp in total length and encompassed 13 protein-coding genes, two ribosomal RNAs, 22 transfer RNAs, and one non-coding region (D-loop). All of the genes were encoded on the H-strand with the exception of ND6 and eight tRNAs, which is a conserved pattern of the mitogenome for other vertebrates. The mitogenome of A. carneipes was dominated by higher AT (56.5%) than GC (43.5%) content. In the resulting phylogenetic tree using complete mitogenome sequences, flesh-footed shearwater and gray petrel (Procellaria cinerea) grouped together despite the high genetic distance (11.0%) between them, belonging to family Procellariidae. However, the phylogenetic tree was consistent with a previous study using partial nucleotide sequences of the cytochrome b gene. These results highlight that further mitogenome sequences will be required from the closely related species under the genus Ardenna to delineate well-resolved phylogenetic classification at the genus and or species level. The present study provides a reference mitochondrial genome of flesh-footed shearwater for further molecular studies.
Introduction
The development of nuclear marker data sets and mitochondrial sequences has provided major advances to phylogenetic analyses. As the mitochondrial genome is maternally inherited and is haploid, its’ effective population size is a quarter that of a nuclear‐autosomal gene (Moore Citation1995). Therefore, the mitochondrial phylogeny has a considerably higher probability of tracking the true species tree, because lineage sorting of mitochondrial haplotypes is more likely to resolve along a given internal branch of the phylogeny than is lineage sorting of nuclear genes (Moore Citation1995). Technical advances have made it easier to obtain complete mitochondrial genome sequences rather than small fragments of the mitochondrial genome. This innovation takes advantage of high-throughput Next-generation sequencing (NGS) techniques to produce high yields of sequencing reads, and sophisticated bioinformatics programs for extracting and assembling the entire mitochondrial genomes from almost any eukaryotic species for which total DNA can be isolated (Smith Citation2016). Here, we use the NGS technology to obtain whole mitochondrial genome of the vulnerable flesh-footed shearwater (Ardenna carneipes; formerly Puffinus carneipes).
The status of the world’s bird population in recent decades has deteriorated, with the largest impact observed in seabird populations (Croxall et al. Citation2012). One such example is the flesh-footed Shearwater (A. carneipes). The bird population of flesh-footed shearwater has been deteriorating for many years and is listed as vulnerable in the state of Western Australia and New South Wales and rare in South Australia (Reid et al. Citation2013; Lavers Citation2014). The species is also listed nationally vulnerable in New Zealand (Robertson et al. Citation2013) and has been recommended for listing under the Agreement on the Conservation of Albatrosses and Petrels (Copper and Baker Citation2008; ACAP Citation2019). However, molecular based studies on the A. carneipes are also very limited, and only partial mitochondrial sequences of this species are available in the NCBI database (Nunn and Stanley Citation1998; Penhallurick and Wink Citation2004; Lombal et al. Citation2018). This work intended to (i) generate and assemble the first mitogenome data of A. carneipes from Australia using a next-generation sequencing platform, and (ii) reveal the phylogenetic relationships of A. carneipes utilizing selected mitogenome sequences available in GenBank.
Materials and methods
Source of sampling and extraction of DNA
A cutaneous tissue sample was collected from a single flesh-footed shearwater (Ardenna carneipes) originating from South of Lord Howe Island in New South Wales (GPS location: 32.53° S, 159.08° E, and collected by Jennifer L. Lavers, E-mail: [email protected]) during April 2015, and was sent to Prof Shane R. Raidal (E-mail: [email protected]) at Charles Sturt University for further analysis (sample voucher CS15-1527, stored at Veterinary Diagnostic Laboratory, Charles Sturt University, Wagga Wagga, New South Wales, Australia) (Sarker et al. Citation2017). Ethics for sample collection was approved by the Lord Howe Island Board (permit no. LHIB 02/14) and the Charles Sturt University and University of Tasmania Animal Ethics Committees (permit no. 09/046, A0010874, and A0011586). The genomic DNA was extracted utilizing a Qiagen Blood and Tissue mini kit (Qiagen, Germany), and stored at −20 °C until further use at Charles Sturt University (Sarker et al. Citation2017).
Sequencing, assembly and annotation of complete mitogenome of A. carneipes
The library preparation and sequencing was performed as previously described (Sarker et al. Citation2017). Briefly, an Illumina paired-end sample preparation kit (Illumina, San Diego, CA) was utilized to generate a paired-end library with an insert size of 150 bp, and sequencing performed by Novogene, China on a HiSeq4000 sequencing platform (Illumina). A previously established pipeline utilizing the Geneious (version 10.2.2, Biomatters, New Zealand) and CLC Genomics Workbench (version 9.5.4) platforms was used for data analysis (Sarker et al. Citation2017; Sarker et al. Citation2019a; Sarker et al. Citation2019b). Briefly, the complete mitochondrial genome of A. carneipes was assembled from a total of 14.42 million reads with a read length of 150 bp. Cleaned unmapped reads were used as input data for de novo assembly using SPAdes assembler (version 3.10.1) (Bankevich et al. Citation2012) in Geneious (version 10.2.2). This resulted in the generation of a 16,370 bp mitogenome obtained from A. carneipes. A total of 13.96 million clean raw reads were mapped back to the mitogenome of A. carneipes that resulted in an average coverage of 65.44x. The default parameter under the genetic code of vertebrate mitochondrial (transl_table 2) in Geneious (version 10.2.2) was utilized for annotation of the sequenced mitogenome of A. carneipes.
Comparative genomics and phylogenetic analysis
The genetic organization of the newly assembled mitogenome of A. carneipes was visualized using Geneious software. The newly assembled mitogenome sequence of A. carneipes together with other 18 selected mitogenome sequences belonging to the order Procellariiformes were utilized to perform phylogenetic analyses, where the D-loop region was manually removed, and approximately 15.0 kbp aligned sequences were used in further analyses. Nucleotide sequences of partial cytochrome b gene were selected from the genus Ardenna. MAFTT (version 7.450) and G-INS-i (gap open penalty 1.53; offset value 0.123) algorithms were implemented in Geneious (version 7.388) to align the nucleotide sequences (Katoh and Standley Citation2013). To determine the best-fit model to compute phylogenetic analyses, a model test was performed using CLC Genomics Workbench (version 9.5.4), which favored a general-time-reversible model with gamma distribution rate variation and a proportion of invariable sites (GTR + G + I). Maximun likelihood (ML) phylogenetic analysis was performed under GTR substitution model with 1000 bootstrap support in Geneious.
Results and discussion
Structure of A. carneipes mitogenome
The assembled mitogenome of flesh-footed shearwaters had a total length of 16,370 bp circular genome (GenBank accession no. MT948200). The overall mitogenome architecture of A. carneipes was mostly conserved compared to other mitogenome sequences in the family Procellariidae (Lounsberry et al. Citation2015; Jung et al. Citation2019). The mitogenome of A. carneipes contained one control region (D-loop), 22 transfer RNAs (tRNAs), two ribosomal RNA (rRNA) encoding regions (12S and 16S rRNA), and 13 protein-coding genes (PCGs) ( and ). The mitochondrial genome structural map of A. carneipes revealed that the majority of the genes were encoded on the heavy strand (H-strand), with only a small handful of genes being encoded on the light strand (L-strand) ( and ).
Figure 1. (A) Gene composition of the mitochondrial genome for A. carneipes is visualized using Geneious (version 10.2.2). Internal frame plot represents GC (blue)/AT (green) content for the mitogenome of A. carneipes (sliding window size: 54). (B) Consensus identity graph representing the mean pairwise identity over all pairs in the column between the mitochondrial genome of A. carneipes (GenBank accession no. MT948200) and Procellaria cinerea (GenBank accession no. AP009191); where, color green: 100% identity, greeny-brown: at least 30% and under 100% identity and red: below 30% identity. Vertical black lines highlighting the SNPs for the mitochondrial genome of A. carneipes compared to mitochondrial genome of Procellaria cinerea. Dark red and blue colored open reading frames correcpondence to rRNA and PCGs, respectively.
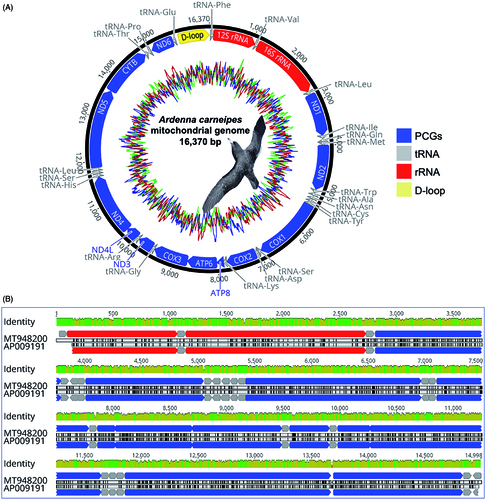
Table 1. Characteristics of the mitochondrial genome of A. carneipes.
The nucleotide composition of the mitogenome of A. carneipes was similar to what has been observed in other vertebrate mitochondrial genomes, with the A + T content being higher (56.50%) than G + C content (43.50%), and guanine having the lowest frequency (A > C > T > G). The size and genomic coordinate of 13 protein-coding genes (PCGs) in A. carneipes mitogenome was consistent with others member of the family Procellariidae (Slack et al. Citation2006; Watanabe et al. Citation2006; Lounsberry et al. Citation2015; Jung et al. Citation2019). A large percentage (69.6%) of the mitogenome was constituted by protein-coding genes (11,394 bp in total length, encoding for 3,684 amino acids, excepting termination codons).
Genetic diversity and phylogenetic analysis
The mitogenome sequence of A. carneipes showed relatively high genetic distance ranging from 0.11 to 0.16 (11.0% to 16.0%) and demonstrated the highest genetic similarity (89.04%) with Procellaria cinerea (GenBank accession no. AP009191). In the absence of any other complete mitogenome sequences in the genus Ardenna, we used mitogenome of P. cinerea to understand the variation and calculate single-nucleotide polymorphisms (SNPs) of A. carneipes. We found that there were 3006 SNPs () in the mitogenome of A. carneipes.
The resulting ML phylogenetic tree delineated seabirds families Procellariidae, Diomedeidae and Hydrobatidae, which formed a well-resolved monophyletic group with high bootstrap support (100%) (). This relationship is in agreement with the results of a previous study (Nunn and Stanley Citation1998). However, the phylogenetic relationship of A. carneipes was not well resolved at the genus level due to the lack of available mitogenome sequences at the genus level. Instead, the newly sequenced mitogenome of A. carneipes formed a well-supported clade with Procellaria cinerea (). Further, large-scale sequencing will be required to delineate well-resolved genus or species level phylogenetic trees. By also building a phylogenetic tree with partial nucleotide sequences of cytochrome b gene selected from the genus Ardenna (), we saw very consistent tree topology that has been established previously (Nunn and Stanley Citation1998). The seabird species A. carneipes clustered with A. creatopus (bootstrap support 95%). The results of the current study based on partial nucleotide sequences of cytochrome b gene are broadly consistent with those of the previous study (Nunn and Stanley Citation1998), but further mitogenome sequencing will be required to delineate well-resolved genus-level phylogenetic classification.
Figure 2. (A) Maximum-likelihood (ML) phylogenetic tree displaying the evolutionary relationship between A. carneipes with other selected species under the order of Procellariiformes based on complete mitogenome. (B) ML phylogenetic tree displaying the evolutionary relationship between A. carneipes with other selected species under the genus Ardenna based on partial cytochrome b gene. The new complete mitogenome of A. carneipes was highlighted by magenta color.

Conclusions
This study reports the first mitogenome of flesh-footed shearwater as a reference for further molecular studies. According to the phylogenetic trees obtained using complete mitogenome sequences, flesh-footed shearwater and gray petrel grouped together despite the high genetic distance (11.0%) between them, belonging to family Procellariidae, which is likely due to low sampling of mitognomes in Procellaridae. Further studies should integrate both morphological data and nuclear and mitogenome sequences from the closely related taxa to delineate well-resolved phylogenetic classification at the genus and or species level.
Disclosure statement
The authors declare no conflicts of interest. The authors alone are responsible for the content and writing of the manuscript.
Data availability statement
The complete mitogenome sequence of A. carneipes genome was deposited in GenBank under the accession number MT948200. Raw sequencing datasets that support the findings of this study are accessible through Mendeley Data repository via the following Link http://dx.doi.org/10.17632/t78p5tgvrc.1 (Sarker et al. Citation2020).
Additional information
Funding
References
- ACAP. 2019. Report of the eleventh meeting of the Advisory Committee. Eleventh Meeting of ACAP’s Advisory Committee; p. 57.
- Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, et al. 2012. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 19(5):455–477.
- Copper J, Baker GB. 2008. FORUM PAPER: identifying candidate species for inclusion within the Agreement on the Conservation of Albatrosses and Petrels. Mar Ornithol. 36:1–8.
- Croxall JP, Butchart SHM, Lascelles BEN, Stattersfield AJ, Sullivan BEN, Symes A, Taylor P. 2012. Seabird conservation status, threats and priority actions: a global assessment. Bird Conserv Int . 22(1):1–34.
- Jung J-W, Lee H, Choi H-G, Kim J-H. 2019. Complete mitogenome of the Cape petrel Daption capense from Barton Peninsula, King George Island, Antarctica. Mitochondrial DNA B. 4(1):1704–1705.
- Katoh K, Standley DM. 2013. MAFFT Multiple Sequence Alignment Software Version 7: improvements in performance and usability. Mol Biol Evol. 30(4):772–780.
- Lavers JL. 2014. Population status and threats to Flesh-footed Shearwaters (Puffinus carneipes) in South and Western Australia. ICES J Mar Sci. 72(2):316–327.
- Lombal AJ, Wenner TJ, Lavers JL, Austin JJ, Woehler EJ, Hutton I, Burridge CP. 2018. Genetic divergence between colonies of Flesh-footed Shearwater Ardenna carneipes exhibiting different foraging strategies. Conserv Genet. 19(1):27–41.
- Lounsberry ZT, Brown SK, Collins PW, Henry RW, Newsome SD, Sacks BN. 2015. Next-generation sequencing workflow for assembly of nonmodel mitogenomes exemplified with North Pacific albatrosses (Phoebastria spp.). Mol Ecol Resour. 15(4):893–902.
- Moore WS. 1995. Inferring phylogenies from mtDNA variation: mitochondrial-gene trees versus nuclear-gene trees. Evolution. 49(4):718–726.
- Nunn GB, Stanley SE. 1998. Body size effects and rates of cytochrome b evolution in tube-nosed seabirds. Mol Biol Evol. 15(10):1360–1371.
- Penhallurick J, Wink M. 2004. Analysis of the taxonomy and nomenclature of the Procellariiformes based on complete nucleotide sequences of the mitochondrial cytochrome b gene. EMU - Austral Ornithol. 104(2):125–147.
- Reid T, Hindell M, Lavers JL, Wilcox C. 2013. Re-examining mortality sources and population trends in a declining seabird: using Bayesian methods to incorporate existing information and new data. PLoS One. 8(4):e58230.
- Robertson HA, Dowding JE, Elliott GP, Hitchmough RA, Miskelly CM, O’Donnell CFJ, Powlesland RG, Sagar PM, Scofield RP, Taylor GA. 2013. Conservation status of New Zealand birds 2012. New Zealand Threat Classification Series 4. Wellington: Department of Conservation.
- Sarker S, Das S, Lavers JL, Hutton I, Helbig K, Imbery J, Upton C, Raidal SR. 2017. Genomic characterization of two novel pathogenic avipoxviruses isolated from pacific shearwaters (Ardenna spp.). BMC Genomics. 18(1):298.
- Sarker S, Raidal S, Lavers J. 2020. Metagenomic dataset of flesh-footed shearwater (Ardenna carneipes). Mendeley Data. V1.
- Sarker S, Sutherland M, Talukder S, Das S, Forwood JK, Helbig K, Raidal SR. 2019a. The first complete mitogenome of Indian ringneck (Psittacula krameri) demonstrates close phylogenetic relationship with Eclectus parrot. Mitochondrial DNA B Resour. 4(2):3579–3581.
- Sarker S, Talukder S, Sutherland M, Forwood JK, Helbig K, Raidal SR. 2019b. Characterization of the first mitochondrial genome of a little Corella (Cacatua sanguinea) and its phylogenetic implications. Mitochondrial DNA B Resour. 4(2):3792–3794.
- Slack KE, Jones CM, Ando T, Harrison GL, Fordyce RE, Arnason U, Penny D. 2006. Early penguin fossils, plus mitochondrial genomes, calibrate avian evolution. Mol Biol Evol. 23(6):1144–1155.
- Smith DR. 2016. The past, present and future of mitochondrial genomics: have we sequenced enough mtDNAs? Brief Funct Genomics. 15:47–54.
- Watanabe M, Nikaido M, Tsuda TT, Kobayashi T, Mindell D, Cao Y, Okada N, Hasegawa M. 2006. New candidate species most closely related to penguins. Gene. 378:65–73.