
Abstract
Scutellaria Linn. is a perennial herb with about 300 species. This genus has high medicinal value and many are used in Traditional Chinese Medicine (TCM). In this study, we sequenced and assembled the complete chloroplast genomes of Scutellaria tsinyunensis and S. tuberifera. Subsequently, we conducted a comprehensive comparative genomics analysis with 12 other published Scutellaria species. These genomes all had a conserved quartile structure, and the gene contents, gene sequences and GC contents are highly similar. The study on the genetic characteristics and nucleotide substitution rate of different genes found that the protein-coding genes of chloroplasts have differed greatly. Most genes are under purifying selection, but the rps12 gene may have undergone positive selection. Besides, we identified three hypervariable regions as potential markers for Scutellaria taxa, which could play an important role in species identification of Scutellaria. Phylogenetic analysis showed that the 14 Scutellaria taxa were divided into two major clades. Moreover, the variation of IR regions is closely related to the evolutionary history as was reconstructed based on SNPs. In conclusion, we provided two high-quality chloroplast reference genomes of Scutellaria, this reliable information and genomic resources are valuable for developing of efficient DNA barcodes as reconstruction of chloroplast evolutionary history of the genus.
1. Introduction
Scutellaria Linn. is a perennial herb of about 300 species, which belongs to the family Lamiaceae. Scutellaria plants are widely distributed throughout the world except for tropical Africa. Several species from Scutellaria are used in Traditional Chinese Medicine (TCM) with the functions of clearing away heat and dampness, purging internal heat, and detoxification (Zhao T et al. Citation2019). For instance, the dried roots of S. baicalensis, also known as ‘Huang Qin’, are used for liver and lung complaints and even used for complementary cancer treatments (EghbaliFeriz et al. Citation2018; Wang CZ et al. Citation2020). Phytochemical studies have shown that the main active compounds of Scutellaria species are a series of flavonoids, include wogonin, wogonoside, baicalin, and baicalein (Wang ZL et al. Citation2018; Zhao Q et al. Citation2019). By now, the research on Scutellaria taxa is mainly focused on chemical composition, medicine activity and biological technology (Wang ZL et al. Citation2018; Zhao Q et al. Citation2019). In particular for S. baicalensis, which is favored for excellent effect in disease treatment. However, the resource identification based on molecular phylogenetic studies is relatively scarce.
Chloroplast genome (referred to as cp genome in the following text) plays an important role in plant photosynthesis (Szabò and Spetea Citation2017) and are widely used in phylogenetic studies and species identification (Santos and Pereira Citation2018; Wang A et al. Citation2018). Due to its conservative genome structure and contents, the cp genome has become an ideal model for evolutionary and comparative genomic studies (Shin et al. Citation2016). Although the cp genome is relative conserved compared to the nuclear genomes, it also contains highly variable regions that were widely used as molecular markers (Liu ML et al. Citation2018; Liu X et al. Citation2018; Pang et al. Citation2019; Thakur et al. Citation2019). For instance, matK, rbcL, and trnH-psbA were used as the universal DNA barcodes for distinguishing species (de Vere et al. Citation2015; Guo et al. Citation2011; Yu et al. Citation2021). In a recent study, Zhao et al. (Citation2020) reported 8 cp genomes of Scutellaria plants, which have greatly enriched the cp genome resources. However, cp genome sequencing is still inadequate in such a moderately large genus, and the comparative genomic analysis of cp genomes is incomplete.
In our study, we have sequenced two cp genomes of Scutellaria species, they are S. tsinyunensis C.Y. Wu & S. Chow and S. tuberifera C. Y. Wu et C. Chen. Among them, S. tsinyunensis is an endangered perennial herb endemic to Mt. Jinyun, Chongqing, China (Li and Hedge Citation1994). Subsequently, we conducted a comprehensive comparative genomics analysis with 12 other published Scutellaria taxa. In particular, we focused on the molecular evolution of chloroplast genomes, such as the expansion/contraction of IR regions, the evolution of protein-coding genes, and the identification of hypervariable regions. The entire cp genome sequences were used as a super-barcode to determine the phylogenetic position of Scutellaria plants.
2. Materials and methods
2.1. Sampling, DNA extraction and sequencing
The fresh leaves of two Scutellaria species, S. tsinyunensis and S. tuberifera were collected from Mt. Jinyun, Chongqing (Geospatial coordinates: N29.842889, E106.394527) and Greenhouse 9, Southwest University, Chongqing (Geospatial coordinates: N29.817767, E106.421054), respectively. The samples have been deposited in the herbarium of Southwest University, Chongqing, China with the accession number: 20200320CQ-1 and 20200320CQ-2, respectively. The total genomic DNA was extracted by using CTAB method (Arseneau et al. Citation2017). The DNA library with an insert size of 350 bp was constructed using the NEBNext® library building kit (Emerman et al. Citation2017) and sequenced by using the Hiseq Xten PE150 sequencing platform. Sequencing produced a total of 4.19 G and 5.23 G raw data. A total of 19,816,746 and 22,736,325 raw reads (2 × 150 bp) were obtained. Clean data were obtained by removing low-quality sequences: sequences with a quality value of Q < 19 accounted for more than 50% of the total base, and sequences with more than 5% bases being ‘N’.
2.2. Genome assembly and annotation
Genome assembly from the clean data was accomplished utilizing NOVOPlasty version 2.7.2 (Dierckxsens et al. Citation2017), with a k-mer length of 39 bp and a sequence fragment of the rbcL gene from maize as the seed sequence. The average base-coverage was 499.3 (S. tsinyunensis) and 506.6 (S. tuberifera). Then, we use Geneious version 8.1 (Auckland, New Zealand) (Kearse et al. Citation2012) to map all clean reads to the assembled genome sequence to verify whether the spliced contigs were correct. The cp genome was annotated initially by using CPGAVAS2 (Shi et al. Citation2019) using the reference dataset of 2544-plastomes. Geseq was then used to confirm the annotation results (Tillich et al. Citation2017). Furthermore, the annotations with problems were manually edited by using Apollo (Misra and Harris Citation2005).
2.3. Sequence analysis and genome comparison
The GC content was conducted by using the cusp program provided by EMBOSS version 6.3.1 (Rice et al. Citation2000). IRscope (https://irscope.shinyapps.io/irapp/) was used for visualizing the IR boundaries in these cp genomes (Amiryousefi et al. Citation2018). A total of 78 orthologous genes and 89 intergenic spacer regions (IGSs) among 14 Scutellaria species were identified and extracted by using Phylosuite version 1.2.1 (Zhang et al. Citation2020). The corresponding nucleotide sequences were aligned by using MAFFT version 7.450 (https://mafft.cbrc.jp/alignment/server/) (Rozewicki et al. Citation2019) implemented in Phylosuite. We used MEGA version 6.0 (Tamura et al. Citation2013) to calculate the percentage of variable sites (PV) in protein-coding genes and the pairwise K2-P distance in IGSs. Then, we used DnaSP version 6.0 (Rozas et al. Citation2017) to calculate the nucleotide diversity (Pi) among the protein-coding sequence.
2.4. Nucleotide substitution rate analysis
The protein-coding sequences in the previous step were processed in parallel. We used the CODEML module in PAML version 4.9 (Yang Citation2007) to estimate rates of nucleotide substitution, including dN (nonsynonymous), dS (synonymous), and the ratio of nonsynonymous to synonymous rates (dN/dS). The detailed parameters were: CodonFreq = 2 (F3 × 4 model); model = 0 (allowing a single dN/dS value to vary among branches); cleandata = 1 (remove sites with ambiguity data); other parameters in the CODEML control file were left at default settings. The phylogeny tree structure of each gene was generated by using the maximum-likelihood (ML) method implemented in RaxML version 8.2.4 (Stamatakis Citation2014).
2.5. Phylogenetic analysis
The cp genome sequences of 14 species belonging to Lamiaceae were downloaded from GenBank (Table S1). Two species (Lamium album and Stachys byzantina) were used as outgroups. A total of 16 complete cp genome sequences were aligned by using MAFFT version 7.450 online version with default setting (Rozewicki et al. Citation2019). These aligned sequences were used to construct the phylogenetic trees by using the ML method implemented in RaxML version 8.2.4 (Stamatakis Citation2014). The parameters were ‘raxmlHPC-PTHREADS-SSE3 -f a -N 1000 -m GTRGAMMA -x 551314260 -p 551314260’. The bootstrap analysis was performed with 1000 replicates.
Table 1. Basic features of the 14 cp genomes from Scutellaria.
3. Results
3.1. General features of cp genomes
The cp genomes of Scutellaria species are characterized by a typical circular DNA molecule with the length of 151,675–152,417 bp. It has a conservative quartile structure which is composed of a LSC region (83,891–84,608 bp), an SSC region (17,305–17,570 bp), and a pair of IR regions (25,208–25,255 bp) (). The GC content analysis showed that the overall GC contents ranged from 38.3% to 38.4% in the 14 cp genomes.
The cp genomes encode a large number of genes. Take S. tsinyunensis for example, the cp genomes comprise 134 genes. Among which, 114 are unique genes, including 80 protein-coding genes, four rRNAs, and 30 tRNAs (). shows the schematic diagram of the cp genomes of S. tsinyunensis. This result is similar to that of other species in this genus (Jiang et al. Citation2017; Lee and Kim Citation2019). In one particular case, two protein-coding genes of S. kingiana, ndhD, and ndhF, are encounter termination codons in advance within the coding frame. As two pseudogenes, they cannot translate the normal protein products. The two genes were not included in subsequent analysis.
Figure 1. Graphic representation of features identified in the cp genomes of Scutellaria plants by using CPGAVAS2. Taking S. tsinyunensis as an example, the map contains four rings. From the center going outward, the first circle shows the forward and reverse repeats connected with red and green arcs, respectively. The next circle shows the tandem repeats marked with short bars. The third circle shows the microsatellite sequences identified using MISA. The fourth circle is drawn using drawgenemap and shows the gene structure on the cp genomes. The genes were colored based on their functional categories, which are shown in the left corner. Label intron-containing genes with *.
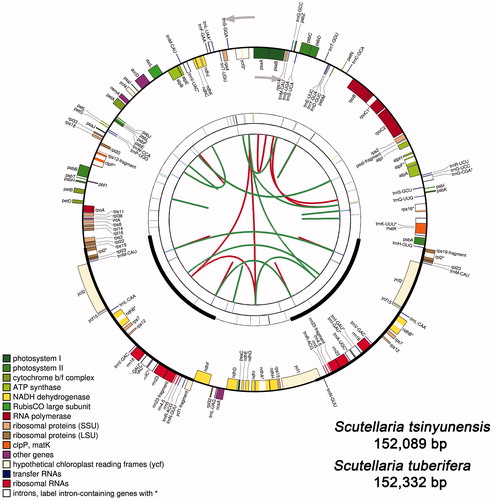
Table 2. Gene contents of the cp genomes in Scutellaria plants.
3.2. Contraction and expansion analysis of IR regions
We observed four genes are span the boundary regions in all 14 species, they are trnH, rps19, ndhF, and ycf1 (). Extensively comparative analysis observed the location of these four genes of Scutellaria species is slightly different. Based on these differences, we divide them into two types (three subtypes). For gene rps19, it overlaps with the IRb regions by 41 bp in type I. However, in type II, the overlap is 46 bp (type IIa) or more than 50 bp (type IIb). For gene ndhF, most sequences are located in SSC regions, it also overlaps with the IRb regions by 32 bp in type I except for S. quadrilobulata (25 bp). In type II, the overlap is 45 bp (type IIa) or 25 bp (type IIb). The variation of ycf1 genes is quite different, and it did not show an obvious classified pattern. It may be related to the high mutation rates of ycf1. It is worth noting that ndhF gene is a pseudogene in S. kinglana.
Figure 2. Comparison of the borders among LSC, SSC, and IR regions of 14 analyzed species. The genes around the borders are shown above or below the main line. The JLB, JSB, JSA, and JLA represent junction sites of LSC/IRb, IRb/SSC, SSC/IRa, and IRa/LSC, respectively.

Interestingly, the ndhF genes cross the border of IRb/SSC, and we observed overlaps of ndhF and the first copy of ycf1. The length of the overlapping regions ranged from 25 to 35 bp in type I and type IIb, but over 120 bp in two species from type IIa, indicating that type I is close to type IIb, and they are quite different from type IIa.
3.3. Genetic characteristics of protein-coding genes
In our study, the Pi value and PV value were highly similar in all 78 genes (). The Pi value (0.0190) and PV value (5.7550) of ycf1 were all the highest. Other genes with high nucleotide polymorphism were rpl32 (0.0176, 5.7471), rps16 (0.0168, 4.9242), and rpl22 (0.0138, 4.1850). The Pi value and PV value were given in parentheses one by one, respectively (Table S2). Five genes (ycf15, petN, psbE, psbN, and rpl23) did not have any variable sites and they are highly conserved.
Figure 3. Genetic characteristics among protein-coding genes of chloroplast in Scutellaria. A. Nucleotide diversity (Pi) and percent variability (PV) of 78 orthologous genes. B. Estimations of nonsynonymous (dN), synonymous (dS) substitution rates, and their ratio (dN/dS) of 78 orthologous genes. A few maximum values were marked with ‘#’.
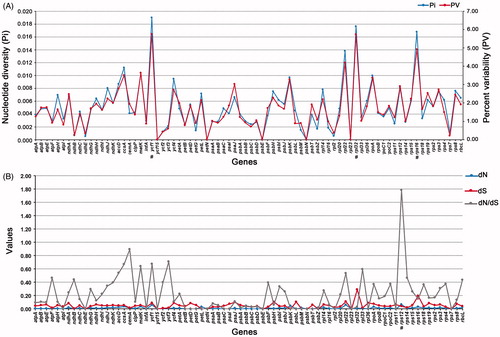
The rates of synonymy (dS) and non-synonymous (dN) substitution rates and their ratios (dN/dS) of 78 orthologous genes were estimated to detect the heterogeneity of substitution rates. Among the 78 genes, rps12, ycf1, rpl22, and psbK had higher dN values, which were 0.0652, 0.0604, 0.0591, and 0.0432, respectively. The dS value of rpl32 was the highest at 0.2896 (, Table S3). The dN/dS value of most genes was less than 0.6, indicating that they have been under purifying selection during evolution. It is worth noting that the dN/dS value of rps12 gene reaches 1.7814, which is likely to undergo positive selection. Other genes with higher dN/dS values are cemA (0.8926), ycf3 (0.7090), ycf1 (0.6734), ccsA (0.6698), and matK (0.6406), which are all active genes in the process of evolution.
Table 3. Mean K2-P distance of 89 IGS of cp genomes from Scutellaria.
3.4. Identification of hypervariable regions
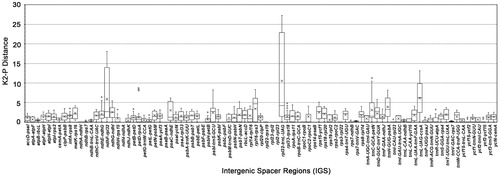
Considering that the protein-coding genes are extremely conserved, we are more focused on the IGSs. As shown in , the K2P distances of the 89 IGSs were quite different. The maximum, minimum and mean K2-P distance showed significant differences in three IGS, which are ndhF-rpl32 (5.8178), trnL-UAG-ccsA (6.1056), and rpl32-trnL-UAG (10.5154). The mean was given in the parentheses, and the details are shown in . The above three IGSs could be used as potential DNA barcodes. Other IGSs with larger differences were trnH-GUG-psbA, rpl16-rps3, trnC-GCA-petN, and psaC-ndhE, which could be used as candidate hypervariable regions.
Figure 4. Boxplot for pairwise comparison of the K2-P distance among 89 intergenic spacers (IGS) of 14 Scutellaria species. The ‘× in each boxplot represents the average K2-P distance. Three IGS had mean K2-P distance over 5, they are ndhF-rpl32 (5.8178), trnL-UAG-ccsA (6.1056) and rpl32-trnL-UAG (10.5154).
3.5. Phylogenetic analysis
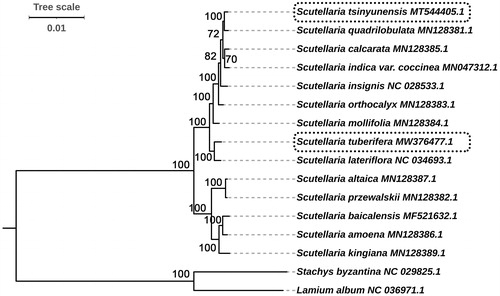
In this study, we selected two outgroups and analyzed the phylogenetic relationships of 14 Scutellaria species. The 16 complete cp genome sequences were used for constructing a ML tree. The phylogenetic trees have high bootstrap support values (100) on most nodes except for three nodes, showing the reliability of the phylogeny recovered (). Our phylogenetic trees displayed two clades clearly, and then further diversified into different subclades. Among the two clades, five species (S. baicalensis, S. Amoena, S. Kingiana, S. Altaica, and S. Przewalskii) were clustered, and the other nine Scutellaria taxa clustered together. In the two species that we sequenced, S. tsinyunensis had the closest relationship with S. quadrilobulata, and S. tuberifera had the closest relationship with S. lateriflora. These results exhibited that the whole cp genome sequences can be used as a super-barcode for species identification with extremely high resolution at the species level.
Figure 5. Phylogenetic relationships of Scutellaria species inferred using maximum-likelihood (ML) method. The phylogenetic tree was constructed using complete cp genome sequences. The number at the bottom of the scale, 0.01, means that the length of the branch represents the replacement frequency of bases at each site of the genome at 0.01. Bootstrap values were calculated from 1000 replicates. Two taxa, namely, Lamium album and Stachys byzantina were used as outgroups.
4. Discussion
Here, we sequenced and assembled the complete chloroplast genomes of S. tsinyunensis and S. tuberifera, which were highly similar to previously published one (Jiang et al. Citation2017; Lee and Kim Citation2019). This result suggested the cp genomes were highly conserved in Scutellaria.
The contraction and expansion of IR regions are considered to be an important reason for the length diversity in cp genomes (Goulding et al. Citation1996). In the comparative analysis, although no significant differences were observed in Scutellaria, we were able to divide the cp genomes into several basic types based on the subtle differences of IR regions. It is worth noting that the dynamic changes of genes near the IR boundary are consistent with the topology of the phylogenetic tree, indicating that, structure variation, e.g. the shift of IR boundaries, reflected similar evolutionary history as was reconstructed based on SNPs.
In previous studies, the evolution of plastid protein-coding genes in Scutellaria was rarely involved. The 14 samples allowed us to conduct a wide range of plastid gene studies in Scutellaria, and we calculated the nucleotide substitution rates to understand the evolution rates of plastid genes in Scutellaria species. We found a wide range of heterogeneous evolutionary rates of the plastid genes. Some genes have higher mutation rates, such as ycf1, rps16, and rpl32. While some are highly conserved and have no mutation sites. This difference in the rates of evolution is important for studying the molecular evolution of chloroplasts, because it is usually related to the purifying or positive selection of genes during evolution. In particular, we observed that the dN/dS value of rps12 gene was greater than 1, suggesting that the gene might have undergone positive selection during evolution processes. As part of the 30S ribosomal subunit, rps12 had the function of rRNA binding, and it is also the only trans-splicing gene in the chloroplast. By contrast, the evolution rate of non-coding regions of the cp genome is generally higher than that of protein-coding regions. The study of non-coding regions or IGSs is helpful for us to find appropriate species-specific DNA barcodes. Based on the results of K2-P distance, we recommended three hypervariable regions, including ndhF-rpl32, trnL-UAG-ccsA, and rpl32-trnL-UAG. These markers could be used to distinguish different species of the genus or even different individuals of the same species.
In summary, our results enrich the data on the cp genomes of Scutellaria and provide the basis for the phylogenetic reconstruction of Scutellaria. We have carried out in-depth studies on plastid genes and deepened our understanding of plastid genes of Scutellaria taxa.
Supplemental Material
Download MS Word (43.5 KB)Acknowledgments
The authors are grateful for the technical support provided by Novogene.
Disclosure statement
The authors declare no conflicts of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
JY conceived and designed this study. SYY and XYP collected the samples and extracted DNA for next-generation sequencing. JLL assembled the complete chloroplast genomes. QLQ, and SYZ annotated and analyzed the chloroplast genome, XYP and YYS wrote the manuscript. JY critically reviewed the manuscript. All authors have read and approved the final version of the manuscript.
Data availability statement
The raw sequencing data and two genome sequences have been deposited in NCBI (https://www.ncbi.nlm.nih.gov/) with accession number: PRJNA680174, MT544405.1 (S. tsinyunensis), and MW376477.1 (S. tuberifera). The sample has been deposited in the herbarium of Southwest University, Chongqing, China with the accession number: 20200320CQ-1 and 20200320CQ-2, respectively.
Additional information
Funding
Related Research Data
References
- Amiryousefi A, Hyvönen J, Poczai P. 2018. IRscope: an online program to visualize the junction sites of chloroplast genomes. Bioinformatics. 34(17):3030–3031.
- Arseneau JR, Steeves R, Laflamme M. 2017. Modified low-salt CTAB extraction of high-quality DNA from contaminant-rich tissues. Mol Ecol Resour. 17(4):686–693.
- de Vere N, Rich TC, Trinder SA, Long C. 2015. DNA barcoding for plants. Methods Mol Biol. 1245:101–118.
- Dierckxsens N, Mardulyn P, Smits G. 2017. NOVOPlasty: de novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 45(4):e18.
- EghbaliFeriz S, Taleghani A, Tayarani-Najaran Z. 2018. Scutellaria: debates on the anticancer property. Biomed Pharmacother. 105:1299–1310.
- Emerman AB, Bowman SK, Barry A, Henig N, Patel KM, Gardner AF, Hendrickson CL. 2017. NEBNext direct: a novel, rapid, hybridization-based approach for the capture and library conversion of genomic regions of interest. Curr Protoc Mol Biol. 119:7.30.31–37.30.24.
- Goulding SE, Olmstead RG, Morden CW, Wolfe KH. 1996. Ebb and flow of the chloroplast inverted repeat. Mol Gen Genet. 252(1–2):195–206.
- Guo X, Wang X, Su W, Zhang G, Zhou R. 2011. DNA barcodes for discriminating the medicinal plant Scutellaria baicalensis (Lamiaceae) and its adulterants. Biol Pharm Bull. 34(8):1198–1203.
- Jiang D, Zhao Z, Zhang T, Zhong W, Liu C, Yuan Q, Huang L. 2017. The chloroplast genome sequence of Scutellaria baicalensis provides insight into intraspecific and interspecific chloroplast genome diversity in Scutellaria. Genes (Basel). 8(9):227.
- Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Drummond A. 2012. Geneious basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 28(12):1647–1649.
- Lee Y, Kim S. 2019. The complete chloroplast genome of Scutellaria indica var. coccinea (Lamiaceae), an endemic taxon in Korea. Mitochondrial DNA Part B. 4(2):2539–2540.
- Li XW, Hedge I. 1994. Flora of China. Vol. 17. Beijing (China); St Louis (MO): Science Press, Missouri Botanical Garden Press.
- Liu ML, Fan WB, Wang N, Dong PB, Zhang TT, Yue M, Li ZH. 2018. Evolutionary analysis of plastid genomes of seven Lonicera L. species: implications for sequence divergence and phylogenetic relationships. IJMS. 19(12):4039.
- Liu X, Zhou B, Yang H, Li Y, Yang Q, Lu Y, Gao Y. 2018. Sequencing and analysis of Chrysanthemum carinatum Schousb and Kalimeris indica. The complete chloroplast genomes reveal two inversions and rbcL as barcoding of the vegetable. Molecules. 23(6):1358.
- Misra S, Harris N. 2005. Using apollo to browse and edit genome annotations. Curr Protoc Bioinformatics. 12(1):9.5.1–9.5.28.
- Pang X, Liu H, Wu S, Yuan Y, Li H, Dong J, Liu Z, An C, Su Z, Li B. 2019. Species identification of oaks (Quercus L., Fagaceae) from gene to genome. IJMS. 20(23):5940.
- Rice P, Longden I, Bleasby A. 2000. EMBOSS: the European molecular biology open software suite. Trends Genet. 16(6):276–277.
- Rozas J, Ferrer-Mata A, Sánchez-DelBarrio JC, Guirao-Rico S, Librado P, Ramos-Onsins SE, Sánchez-Gracia A. 2017. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol Biol Evol. 34(12):3299–3302.
- Rozewicki J, Li S, Amada KM, Standley DM, Katoh K. 2019. MAFFT-DASH: integrated protein sequence and structural alignment. Nucleic Acids Res. 47(W1):W5–W10.
- Santos C, Pereira F. 2018. Identification of plant species using variable length chloroplast DNA sequences. Forensic Sci Int Genet. 36:1–12.
- Shi L, Chen H, Jiang M, Wang L, Wu X, Huang L, Liu C. 2019. CPGAVAS2, an integrated plastome sequence annotator and analyzer. Nucleic Acids Res. 47(W1):W65–W73.
- Shin DH, Lee JH, Kang SH, Ahn BO, Kim CK. 2016. The complete chloroplast genome of the Hare’s ear root, Bupleurum falcatum: its molecular features. Genes (Basel). 7(5):20.
- Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 30(9):1312–1313.
- Szabò I, Spetea C. 2017. Impact of the ion transportome of chloroplasts on the optimization of photosynthesis. J Exp Bot. 68(12):3115–3128.
- Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. 2013. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol. 30(12):2725–2729.
- Thakur VV, Tiwari S, Tripathi N, Tiwari G. 2019. Molecular identification of medicinal plants with amplicon length polymorphism using universal DNA barcodes of the atpF-atpH, trnL and trnH-psbA regions. 3 Biotech. 9(5):188.
- Tillich M, Lehwark P, Pellizzer T, Ulbricht-Jones ES, Fischer A, Bock R, Greiner S. 2017. GeSeq - versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 45(W1):W6–W11.
- Wang A, Wu H, Zhu X, Lin J. 2018. Species identification of Conyza bonariensis assisted by chloroplast genome sequencing. Front Genet. 9:374.
- Wang C-Z, Zhang C-F, Luo Y, Yao H, Yu C, Chen L, Yuan J, Huang W-H, Wan J-Y, Zeng J. 2020. Baicalein, an enteric microbial metabolite, suppresses gut inflammation and cancer progression in Apc(Min/+) mice. Clin Transl Oncol. 22(7):1013–1022.
- Wang ZL, Wang S, Kuang Y, Hu ZM, Qiao X, Ye M. 2018. A comprehensive review on phytochemistry, pharmacology, and flavonoid biosynthesis of Scutellaria baicalensis. Pharm Biol. 56(1):465–484.
- Yang Z. 2007. PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol. 24(8):1586–1591.
- Yu J, Wu X, Liu C, Newmaster S, Ragupathy S, Kress WJ. 2021. Progress in the use of DNA barcodes in the identification and classification of medicinal plants. Ecotoxicol Environ Safety. 208:111691.
- Zhang D, Gao F, Jakovlic I, Zou H, Zhang J, Li WX, Wang GT. 2020. PhyloSuite: an integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol Ecol Resour. 20(1):348–355.
- Zhao F, Li B, Drew BT, Chen Y-P, Wang Q, Yu W-B, Xiang C-L. 2020. Leveraging plastomes for comparative analysis and phylogenomic inference within Scutellarioideae (Lamiaceae). PLoS One. 15(5):e0232602.
- Zhao Q, Yang J, Cui MY, Liu J, Fang Y, Yan M, Martin C. 2019. The reference genome sequence of Scutellaria baicalensis provides insights into the evolution of wogonin biosynthesis. Mol Plant. 12(7):935–950.
- Zhao T, Tang H, Xie L, Zheng Y, Ma Z, Sun Q, Li X. 2019. Scutellaria baicalensis Georgi. (Lamiaceae): a review of its traditional uses, botany, phytochemistry, pharmacology and toxicology. J Pharm Pharmacol. 71(9):1353–1369.