
Abstract
Haemaphysalis longicornis (Ixodida: Ixodidae), the Asian longhorned tick, which is native to temperate East Asia, has been recently detected in the northeastern region of the United States, drawing concerns about its potential impact on the US animal and public health sectors. Knowledge about the genetic features of H. longicornis found in the US is limited. Therefore, we sequenced the complete mitochondrial genome (mt-genome) from two H. longicornis ticks recently collected in the State of New York, USA, in 2020. These ticks were morphologically identified and tested for tick-borne pathogens at the Connecticut Veterinary Medical Diagnostic Laboratory (Storrs, CT). The mt-genome was 14,694 bp in length and encoded 37 genes, including 13 protein-coding genes, 22 transfer RNAs, and two ribosomal RNAs. Phylogenetic analysis showed that the mt-genome clustered with those of other H. longicornis identified in China. The mt-genome sequence was 99.7% identical to a H. longicornis mt-genome (GenBank: MK439888) collected in China. The cox1 gene haplotype in these ticks belonged to the H1 type, which is the dominant haplotype present in central NJ and Staten Island, NY. The complete mt-genome data are needed to provide insights into genetic changes and phylogenetic studies of H. longicornis ticks.
In Asia, Haemaphysalis longicornis Neumann (Ixodida: Ixodidae), the Asian longhorned tick, is considered a primary vector for the Severe Fever with Thrombocytopenia Syndrome virus (SFTSV) (Liu et al. Citation2014). These ticks also are competent vectors for the Spotted Fever Group Rickettsia spp (Zou et al. Citation2011) and Rickettsia japonica causing Japanese spotted fever, a fetal zoonotic disease (Noguchi et al. Citation2018). In addition, other tick-borne pathogens including Anaplasma, Borrelia, and Ehrlichia species have also been detected in H. longicornis (Sun et al. Citation2008; Egizi et al. Citation2020). While H. longicornis is indigenous to East Asia and southeast Russia (Beard et al. Citation2018; Egizi et al. Citation2020), it has also been found in the U.S. It was initially identified in 2017 on a sheep in New Jersey, and since then, the species has been reported in 15 states. Recent reports of heavy H. longicornis infestations on cattle (Bos taurus) and white-tailed deer (Odocoileus virginianus) together with wide distribution of this invasive species have drawn concern about its potential impact on US livestock and public health sectors (Egizi et al. Citation2020). Considering its importance, there is limited knowledge on the genetic features of H. longicornis found in the U.S.
Mitochondrial (mt)-genomes are characterized by their simple structure, small molecular weight, maternal inheritance, relatively high mutation rates, and the lack of recombination (Wang et al. Citation2019a, Citation2019b). With these properties, molecular approach using mitochondrial genome (mt-genome) has been recently used for identification and characterization of ticks as well as molecular evolution, phylogeny, and genealogy of ticks (Liu et al. Citation2013, Citation2020; Cameron Citation2014; Geng et al. Citation2017; Wang et al. Citation2019a, Citation2019b; Egizi et al. Citation2020). As of 11 February 2021, only five complete mt-genome sequences have been reported in NCBI GenBank database, mainly originated from China. In the present study, we report the first complete mt-genome sequence of two H. longicornis ticks found during the end of June in 2020 in the state of New York, U.S.
In June 2020, two tick specimens collected from Rockland County, NY (latitude 41.147594, and longitude −73.989304) were submitted to the Connecticut Veterinary Medical Diagnostic Laboratory (CVMDL, Holly McGinnis, [email protected], Maureen Sims, [email protected]), University of Connecticut for identification (speciation) and testing for tick-borne pathogens. The tick 1 (20-2319) was identified as an adult female H. longicornis tick and the tick 2 (20-2320) as a nymph stage H. longicornis using the morphological key of previous study (Egizi et al. Citation2019). These moderately engorged ticks were tested by SYBR green based qPCR for tick-borne pathogens (Pietila et al. Citation2000; Adelson et al. Citation2004; Pesquera et al. Citation2015; Muhammad et al. Citation2019). The tick 2 was positive for Anaplasma phagocytophilum. DNA was extracted from ticks using a Nucleospin Tissue kit (Macherey-Nagel, Bethlehem, PA), and genome sequencing was performed on MiSeq sequencer (Illumina, San Diego, CA). Quality-filtered reads (Q > 30 and minimum length >100) by BBduk (https://sourceforge.net/projects/bbmap) were mapped to the reference sequence (GenBank accession no. MK450606) using Bowtie 2 (Langmead and Salzberg Citation2012). Consensus sequences were annotated by MITOS (Bernt et al. Citation2013).
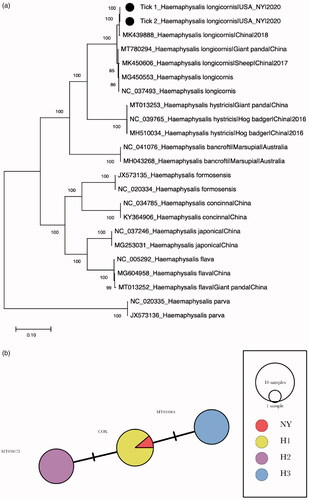
The complete mt-genome sequences of all Haemaphysalis ticks were retrieved from the GenBank for phylogenetic analysis (n = 22). The sequences were aligned using MAFFT (https://mafft.cbrc.jp/alignment/software/). The maximum-likelihood (ML) phylogeny was constructed using RAxML-v8 (Stamatakis Citation2014) using the GTR + G4 substitution model with 1000 rapid bootstrapping. We identified the cox1 haplotype of two samples. Egizi et al. (Citation2020) showed that three cox1 haplotypes (H1–3) of H. longicornis were detected within the U.S. A minimum spanning haplotype network was created using PopART 1.7 (Leigh and Bryant Citation2015) with eight randomly selected cox1 sequences of each haplotype (Egizi et al. Citation2020).
The mt-genome sequences of two ticks obtained in this study were identical. The mt-genome was 14,694 bp in length and classified as type III mt-genome according to the previous study (Montagna et al. Citation2012). It encoded 13 protein-coding genes (PCGs) (cox1–3, nd1–6, nd4L, cob, atp6, and atp8), 22 tRNA genes, two rRNA genes including 16S rRNA (1205 bp) and 12S rRNA (767 bp), and two noncoding regions (NCRs) located at rrnS-trnI and trnL1-trnC. A tandem repeat was found in apt8 gene using Tandem Repeat Finder (Benson Citation1999). The nucleic acid base content was 38.3% A, 13.0% C, 9.7% G, and 39.0% T. ATT start codon was used by nd1, nd2, nd3, nd4L, nd5, cox1, and cox2. ATG codon was used by atp6, cob, and nd4, ATA codon by atp8 and cox3, and ATC codon by nd6. The most PCGs were terminated by TAA stop codon except nd3 and cob that use TAG and nd6 uses the single T, as the stop codon. Among the 37 genes, nine PCGs and 14 tRNAs were located on the forward strand (H-strand), while the remaining genes were transcribed on the reverse strand (L-strand).
The phylogenetic analysis showed that the H. longicornis mt-genome formed a monophyletic cluster with other H. longicornis which is well-supported by high bootstrap value (). The mt-genome sequence was 99.7% identical to a H. longicornis collected in China on 20 March 2018 (GenBank accession no. MK439888). The minimum spanning tree showed that the mt-genome sequences belong to the H1 haplotype which is the dominant in central New Jersey and Staten Island, New York ().
Figure 1. Phylogenetic analysis of the complete mt-genome Haemaphysalis longicornis of this study. (a) Maximum-likelihood phylogeny of complete H. longicornis mt-genomes. The percentages of the bootstrap test in 1000 replicates were shown above the branches. The H. longicornis mt-genome sequenced this study are indicated with black circles. Scale bar indicates nucleotide substitutions per site. (b) Minimum spanning network of H. longicornis cox1 haplotypes identified in the United States. Eight H. longicornis cox1 sequences per haplotype occurring in the United States (H1, H2, and H3) and the H. longicornis cox1 sequence of this study are included in the analysis.
The widespread and establishment of this invasive species in the U.S. highlights the risk to public health since the invasive ticks are capable of transmitting pathogens of human and veterinary concern. The complete mt-genome of H. longicornis found in the U.S. would provide important information to understand genetic diversity and epidemiology of this invasive tick species.
Disclosure statement
The authors report no conflicts of interest.
Data availability statement
The genome sequence data that support the findings of this study are available in GenBank of NCBI at https://www.ncbi.nlm.nih.gov/ under the accession no. MW602986. The associated BioProject number is PRJNA705377, and Bio-Sample numbers are SAMN18086111 and SAMN18086112. The NGS library are stored at the CVMDL, University of Connecticut.
Additional information
Funding
References
- Adelson ME, Rao RV, Tilton RC, Cabets K, Eskow E, Fein L, Occi JL, Mordechai E. 2004. Prevalence of Borrelia burgdorferi, Bartonella spp., Babesia microti, and Anaplasma phagocytophila in Ixodes scapularis ticks collected in Northern New Jersey. J Clin Microbiol. 42:2799–2801.
- Beard CB, Occi J, Bonilla DL, Egizi AM, Fonseca DM, Mertins JW, Backenson BP, Bajwa WI, Barbarin AM, Bertone MA, et al. 2018. Multistate infestation with the exotic disease-vector tick Haemaphysalis longicornis – United States, August 2017–September 2018. MMWR Morb Mortal Wkly Rep. 67:1310–1313.
- Benson G. 1999. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27:573–580.
- Bernt M, Donath A, Juhling F, Externbrink F, Florentz C, Fritzsch G, Putz J, Middendorf M, Stadler PF. 2013. MITOS: improved de novo metazoan mitochondrial genome annotation. Mol Phylogenet Evol. 69:313–319.
- Cameron SL. 2014. Insect mitochondrial genomics: implications for evolution and phylogeny. Annu Rev Entomol. 59:95–117.
- Egizi A, Bulaga-Seraphin L, Alt E, Bajwa WI, Bernick J, Bickerton M, Campbell SR, Connally N, Doi K, Falco RC, et al. 2020. First glimpse into the origin and spread of the Asian longhorned tick, Haemaphysalis longicornis, in the United States. Zoonoses Public Health. 67:637–650.
- Egizi AM, Robbins RG, Beati L, Nava S, Vans CR, Occi JL, Fonseca DM. 2019. A pictorial key to differentiate the recently detected exotic Haemaphysalis longicornis Neumann, 1901 (Acari, Ixodidae) from native congeners in North America. ZooKeys. 818:117–128.
- Geng J, Zheng A, Zou Z, Zhang X. 2017. The complete mitochondrial genome and phylogenetic analysis of Haemaphysalis longicornis Neumann (Acari: Ixodidae). Mitochondrial DNA B Resour. 2:856–857.
- Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods. 9:357–359.
- Leigh JW, Bryant D. 2015. popart: full-feature software for haplotype network construction. Methods Ecol Evol. 6(9):1110–1116.
- Liu GH, Chen F, Chen YZ, Song HQ, Lin RQ, Zhou DH, Zhu XQ. 2013. Complete mitochondrial genome sequence data provides genetic evidence that the brown dog tick Rhipicephalus sanguineus (Acari: Ixodidae) represents a species complex. Int J Biol Sci. 9:361–369.
- Liu Q, He B, Huang SY, Wei F, Zhu XQ. 2014. Severe fever with thrombocytopenia syndrome, an emerging tick-borne zoonosis. Lancet Infect Dis. 14:763–772.
- Liu Y, Wang L, Wang L, Deng L, Wei M, Wu K, Huang S, Li G, Huang Y, Zhang H, et al. 2020. Characterization of the complete mitogenome sequence of the giant panda tick Haemaphysalis hystricis. Mitochondrial DNA B Resour. 5:1191–1193.
- Montagna M, Sassera D, Griggio F, Epis S, Bandi C, Gissi C. 2012. Tick-box for 3′-end formation of mitochondrial transcripts in Ixodida, basal chelicerates and Drosophila. PLoS One. 7:e47538.
- Muhammad J, Rabbani M, Shabbir MZ, Muhammad K, Ghori MT, Chaudhry HR, Ul Hassnain Z, Jamil T, Abbas T, Chaudhry MH, et al. 2019. Cross sectional study and risk factors analysis of Francisella tularensis in soil samples in Punjab Province of Pakistan. Front Cell Infect Microbiol. 9:89.
- Noguchi M, Oshita S, Yamazoe N, Miyazaki M, Takemura YC. 2018. Important clinical features of Japanese spotted fever. Am J Trop Med Hyg. 99:466–469.
- Pesquera C, Portillo A, Palomar AM, Oteo JA. 2015. Investigation of tick-borne bacteria (Rickettsia spp., Anaplasma spp., Ehrlichia spp. and Borrelia spp.) in ticks collected from Andean tapirs, cattle and vegetation from a protected area in Ecuador. Parasit Vectors. 8:46.
- Pietila J, He Q, Oksi J, Viljanen MK. 2000. Rapid differentiation of Borrelia garinii from Borrelia afzelii and Borrelia burgdorferi sensu stricto by LightCycler fluorescence melting curve analysis of a PCR product of the recA gene. J Clin Microbiol. 38:2756–2759.
- Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 30:1312–1313.
- Sun J, Liu Q, Lu L, Ding G, Guo J, Fu G, Zhang J, Meng F, Wu H, Song X, et al. 2008. Coinfection with four genera of bacteria (Borrelia, Bartonella, Anaplasma, and Ehrlichia) in Haemaphysalis longicornis and Ixodes sinensis ticks from China. Vector Borne Zoonotic Dis. 8:791–795.
- Wang T, Zhang S, Pei T, Yu Z, Liu J. 2019a. The complete mitochondrial genome and expression profile of mitochondrial protein-coding genes in the bisexual and parthenogenetic Haemaphysalis longicornis. Front Physiol. 10:982.
- Wang T, Zhang S, Pei T, Yu Z, Liu J. 2019b. Tick mitochondrial genomes: structural characteristics and phylogenetic implications. Parasit Vectors. 12:451.
- Zou Y, Wang Q, Fu Z, Liu P, Jin H, Yang H, Gao H, Xi Z, Liu Q, Chen L. 2011. Detection of spotted fever group Rickettsia in Haemaphysalis longicornis from Hebei Province, China. J Parasitol. 97:960–962.