
Abstract
Hydrangea strigosa Rehder is a wild flowering shrub with high ornamental value. The complete chloroplast genome sequence of H. strigosa was characterized from Hiseq (Illumina Co., San Diego, CA) sequencing data. The chloroplast genome of H. strigosa is 157,905 bp in length with a pair of inverted repeats (IRs) (26,127 bp) which are separated by a large single-copy (LSC) (86,897 bp) and a small single-copy regions (SSC) (18,754 bp). It contains 131 genes, including 85 protein-coding genes, 38 tRNAs genes, and 8 rRNAs genes. The overall GC content of the whole genome is 37.80%. The maximum-likelihood phylogenetic analysis with the complete chloroplast genomes sequence of 22 species of Hydrangeaceae showed that H. strigosa is closely related to H. davidii.
Hydrangea, originates from East Asia, has a long history of use as an ornamental garden plant in temperate regions (Nashima et al. Citation2021; Yoshida et al. Citation2021). It is widely known that the color of Hydrangea sepal changes in the cultivating conditions (Yoshida et al. Citation2021), the sepal color is blue in acidic soil and red in alkaline soil. In addition, Hydrangea has strong ecological adaptability and heavy metal pollution resistance, it is also a hyperaccumulator of aluminum, which can be used as environmental remediation plant (Chen et al. Citation2020). Species circumscription and identification is notoriously difficult in the genus Hydrangea (Smet et al. Citation2017). Based on the ovary position, capsule tips and petal clutch characteristics, Hydrangea is divided into two sections, Sect. Hydrangea and Sect. Calyptranthe. The former can be divided into three series, namely Petalanthae, Heteromallae, and Piptopetalae (Cheng Citation1985). SRAP molecular markers showed that Hydrangea strigosa Rehder belonged to Ser. Piptopetalae (Chen and Peng Citation2012). Hydrangea strigosa is a 1–3 m tall deciduous shrub, and have a wide geographical distribution, dense to sparse forests or thickets in valleys, trail sides on mountain slopes (Wei Citation1995; Smet et al. Citation2017). Because of its large inflorescence, H. strigosa is a wild flowering shrub with high ornamental value. The study for H. strigosa has been focused on seedling technique and physiological characteristics (Chen et al. Citation2020; Li et al. Citation2020), there was no record of complete chloroplast genome sequence to date. In this study, we characterized a complete chloroplast genome of H. strigosa and confirmed the phylogenetic relationship of the genus, to provide genetic information for further research on phylogeography, genetic diversity and evolution.
Fresh leaves of H. strigosa were collected from Baijixun Township, Weixi County, Diqing Tibetan Autonomous Prefecture, Yunnan Province, China (99°1′32.67″E, 27°29′30.1″N). Voucher specimen (SWFU20200713MFY) was deposited in the Herbarium of Southwest Forestry University, China. Total genomic DNA was extracted from silica gel dried leaf tissues using a modified CTAB method (Doyle and Doyle Citation1987). Short-insert library (insert size, 300 bp) was prepared and then sequenced using the Illumina HiSeq 2500-PE150 platform (Illumina, San Diego, CA). The clean reads was obtained from filtered raw reads using NGS QC Toolkit version 2.3.3 with default parameters (Patel and Jain Citation2012). The plastome was de novo assembled by NOVOPlasty (Dierckxsens et al. Citation2017) and annotated by Geneious 20.0.3 (Kearse et al., Citation2012) with the complete chloroplast genome sequence of H. davidii (NC_050783) as the reference genome. The complete chloroplast genome was submitted to GenBank with Accession no. MW218933.
The complete chloroplast genome of H. strigosa is 157,905 bp in length with a quadripartite structure, including a large single-copy region of 86,897 bp, a small single-copy region of 18,754 bp, and two inverted repeat (IR) regions of 26,127 bp. The overall GC content is 37.80%, and the corresponding values of the LSC, SSC, and IR regions are 36.0%, 31.7%, and 43.1%, respectively. The genome encodes 131 genes, including 85 protein-coding genes (PCGs), 38 transfer RNA genes (tRNAs), and 8 ribosomal RNA genes (rRNAs). A total of 54 SSRs were discovered by the online software MISA-web (Beier et al. Citation2017). Among them, the numbers of mono-, di-, tri-, tetra- and penta-nucleotides SSRs are 45, 2, 2, 3, and 2, respectively.
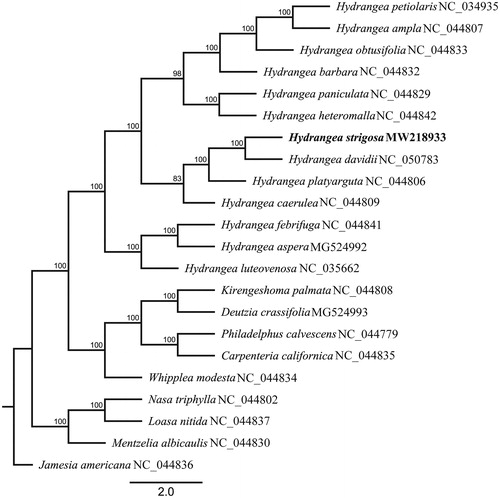
To determine the phylogenetic location of H. strigosa, we used the complete chloroplast genomes sequence of 22 species of Hydrangeaceae including 13 Hydrangea species to construct phylogenetic tree. All cp genome sequences were aligned using MAFFT (Katoh and Standley Citation2013) with default parameters, and then the maximum likelihood tree was constructed using RAxML version 8.2.11 (Stamatakis Citation2014) in which the GTR + G DNA substitution model was selected as a best-fit model using PartitionFinder2 (Lanfear et al. Citation2016), all branch nodes were calculated under 1000 bootstrap replicates. The phylogenetic analysis revealed that all species of Hydrangea were formed one monophyletic clade. Phylogenetic tree showed that H. strigosa was sister to H. davidii with strongly supported under current sampling ().
Figure 1. Maximum-likelihood phylogenetic tree reconstructed by RAxML based on complete chloroplast genome sequences from 13 Hydrangea species and 9 other species of Hydrangeaceae. Numbers on branches are bootstrap support values.
Disclosure statement
No potential conflict of interest was reported by the authors.
Data availability statement
The data that support the findings of this study are openly available in GenBank at https://www.ncbi.nlm.nih.gov/, reference number [MW218933] [SRR12989400], or obtain from the corresponding author.
Additional information
Funding
References
- Beier S, Thiel T, Münch T, Scholz U, Mascher M. 2017. MISA-web: a web server for microsatellite prediction. Bioinformatics. 33(16):2583–2585.
- Chen S, Li YH, Zhao B. 2020. Study on tissue culture and rapid propagation of two wild Hydrangea. Seed. 39(4):60–63. 70.
- Chen HX, Peng JH. 2012. Genetic relationships of partial Hydrangea in China revealed by SRAP markers. Mol Plant Breed. 10(15):1115–1121.
- Cheng WC. 1985. Sylva Sinica. Beijing (China): Chinese Forestry Publishing House. p. 1543–1555.
- Dierckxsens N, Mardulyn P, Smits G. 2017. NOVOPlasty: de novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 45(4):e18.
- Doyle JJ, Doyle JL. 1987. A rapid DNA isolation procedure for small amounts of fresh leaf tissue. Phytochem Bull. 19(1):11–15.
- Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 30(4):772–780.
- Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, et al. 2012. Geneious basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 28(12):1647–1649.
- Lanfear R, Frandsen PB, Wright AM, Senfeld T, Calcott B. 2016. PartitionFinder 2: new methods for selecting partitioned models of evolution for molecular and morphological phylogenetic analyses. Mol Biol Evol. 34(3):772–773.
- Li YH, Chen S, Zhao B. 2020. Effect of aluminum stress on growth and physiological characteristics of Hydrangea tissue culture seedlings. J Zhejiang A&F Univ. 37(6):1064–1070.
- Nashima K, Shirasawa K, Ghelfi A, Hirakawa H, Isobe S, Suyama T, Wada T, Kurokura T, Uemachi T, Azuma M, et al. 2021. Genome sequence of Hydrangea macrophylla and its application in analysis of the double flower phenotype. DNA Res. 28(1):1–10.
- Patel RK, Jain M. 2012. NGS QC toolkit: a toolkit for quality control of next generation sequencing data. PLoS One. 7(2):e30619.
- Smet YD, Clerck OD, Uemachi T, Mendoza CG, Wanke S, Goetghebeur P, Samain MS. 2017. Multilocus coalescent species delimitation to evaluate traditionally defined morphotypes in Hydrangea sect. Asperae (Hydrangeaceae). Mol Phylogenet Evol. 114:415–425.
- Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 30(9):1312–1313.
- Wei ZF. 1995. Hydrangea. Flora of China. Beijing (China): Science Press. p. 411–422.
- Yoshida K, Ito D, Miki N, Kondo T. 2021. Single-cell analysis clarifies mosaic color development in purple hydrangea sepal. New Phytol. 229(6):3549–3557.