
Abstract
The Pacific limpet Cellana nigrolineata is one of the most commonly found limpets in the intertidal shores of Japan. Here, we report the full mitogenome sequence of an individual specimen of the species, which was collected from the intertidal rocky beach in the Nada beach of Gobo City, Wakayama, Japan (33.8316 N, 135.1751 E), in 2018. The sequence was determined by the shotgun sequencing method using the NGS Illumina MiSeq platform. The genomic structure of C. nigrolineata is the same as the previously reported congener, C. radiata, which shows a representative Nacellidae and metazoan mitogenomic structures. The mitogenome has all of its 37 genes included in its 16,153 bp, with one control region located between the tRNA-Cys and tRNA-Gly genes. In order to clarify the phylogenetic position of C. nigrolineata in Gastropoda, a data set including the mitogenomes of 10 patellogastropods, 10 non-patellogastropod gastropods, and four outgroups were used in maximum likelihood inferences. Although with some exceptions, the resulting phylogeny supported the monophylies of traditionally accepted gastropod subclasses, and thus confirms the position of C. nigrolineata in Patellogastropoda.
The limpet Cellana nigrolineata Reeve, 1854, is a true limpet of the family Nacellidae (Patelloidea, Patellogastropoda). It is one of the most commonly present limpet species in the intertidal rocky shores of Japan (Powell Citation1973). The species shows two color variants, which correlates to intraspecific genetic variation (Nakano et al. Citation2010). In this study, we report the full mitochondrial genome (mitogenome) sequence of this species.
A life specimen was collected from the intertidal rocky shore of the Nada Coasts, Gobo City, Wakayama Prefecture, in southwest Japan (33.8316 N, 135.1751 E). The specimen was preserved in 95% ethanol, and then vouchered at The University Museum of The University of Tokyo, Japan (Voucher No. UMUT RM33392). A small piece of tissue (ca. 20 mg) was collected from the mantle tissue, and total genomic DNA was extracted using standard CTAB-Phenol/Chloroform protocol. A sequencing library was prepared using the QiaSeq FX DNA Library Kit (QIAGEN). Fragment size and quality checks were conducted using the TapeStation (Agilent Technologies). The DNA library was then applied into a flow cell of MiSeq Reagent Nanokit V2 (300 cycles) (Illumina), and sequenced on a MiSeq Illumina Next Generation Sequencer. Obtained sequence fragments were edited and assembled into a circular contig using CLC Genomics Workbench ver. 12 (QIAGEN) under default settings. The obtained contig was annotated using the MITOS web server (Bernt et al. Citation2013), which result was further confirmed by manual BLAST searches (Altschul et al. Citation1990).
The newly obtained full mitochondrial genome of Cellana nigrolineata was 16,153 bases-long (Genbank accession number: LC600801), with the GC content 35.44%. The genomic structure is as follows: (1) There are 13 protein-coding, two rRNA, and 22 tRNA genes; (2) There is one control region, located between the tRNA-Cys and tRNA-Gly genes (579 bp-long); (3) Seven of the 13 protein-coding genes are coded on the H chain (ATP6, ATP8, COI, COII, COIII, ND2, ND3); (4) Both the SSU-rRNA and LSU-rRNA coding gene are located on the L chain; (5) 10 tRNA genes (tRNA-Gly, tRNA-Glu, tRNA-Arg, tRNA-Asn, tRNA-Ala, tRNA-Lys, tRNA-Ile, tRNA-Ser (agc), tRNA-Asp, and tRNA-Thr) are located on the H chain.
A maximum likelihood (ML) phylogenetic analysis (Yang Citation1994) was conducted in order to confirm the phylogenetic position of C. nigrolineata among the gastropods. We included the mitogenome sequences of ten patellogastropods, four caenogastropods, two heterobranchias, two neritimorphs, and two vetigastropods, besides three polyplacophorans and an octopod as outgroups in our analysis. The topology of the ML tree with the bootstrap supports is shown in . Detailed explanation of the method is described in the legend of .
Figure 1. Phylogenetic tree showing the affinity of Cellana nigrolineata with its congener, C. radiata, in a monophyletic Patellogastropoda. The bootstrap supports (%) are shown on each node, but 100% support values are not shown. Phylogenetic analyses were conducted on the data matrix (11,407 positions) including all concatenated nucleotide sequences of the mitogenomes, with the third codon positions of the protein-coding genes excluded. Gene sequences were aligned individually using the online version of MAFFT under default settings (Katoh and Standley Citation2013). Aligned sequences were individually edited using the online version of GBlocks using the least stringent settings (Castresana Citation2000). Partitioned ML analyses (four partitions: 1st codon, 2nd codon, rRNA, tRNA) were performed with RAxML-GUI ver. 1-5b1 (Stamatakis Citation2014; Silvestro and Michalak Citation2012), with the GTR + Γ nucleotide substitution model (Yang Citation1994). The rapid bootstrap analyses were conducted with 1000 replications, with four threads running in parallel.
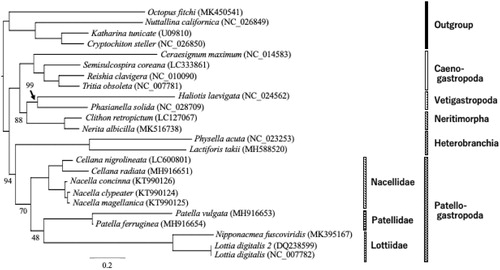
Our phylogeny supports the monophylies of the living gastropods and the historically accepted subclasses sampled in this study (Patellogastropoda, BS: 70; Vetigastropoda, BS: 99; Heterobranchia, BS: 100; Caenogastropoda, BS: 100; and Neritimorpha, BS: 100). However, our tree did not recover Psilogastropoda (Patellogastropoda + Vetigastropoda) (Cunha and Giribet Citation2019), or Orthogastropoda (Ponder and Lindberg Citation1997). In our tree, Patellogastropoda formed a clade with Heterobranchia (BS: 94), which is most likely an artifact caused by long branch attraction (Stöger and Schrödl Citation2013; Williams et al. Citation2014; Feng et al. Citation2020).
Our phylogeny showed the affinity of C. nigrolineata to its congener, C. radiata (BS: 100), and together with the monophyletic Nacella (BS: 100), form a monophyletic Nacellidae (BS: 100). Our tree, however, put Nacellidae as sister to the other two monophyletic families (Patellidae (BS: 100) + Lottiidae (BS: 100)) (BS: 48) sampled in this study. This is incongruent with the previous study of Nakano and Ozawa (Citation2007), which placed Nacellidae and Lottiidae together with Patellidae as the sister group. However, the low statistical supports in our tree suggest that our phylogeny must be considered very carefully. In this study, we did not cover the actual diversity of Patellogastropoda since only 10 species of three families are included in our analyses. Taxon sampling coverage is an important factor to be considered in inferring phylogenies, since the number of taxa affects tree topologies (Pollock et al. Citation2002; Hillis et al. Citation2003; Heath et al. Citation2008), including in Patellogastropoda (Uribe et al. Citation2019). Therefore, future studies on Patellogastropoda phylogeny using mitogenomes must include a more comprehensive taxon sampling, in order to provide an accurate and robust picture of the group’s systematics and evolution (Nakano and Sasaki Citation2011). We are confident, however, that our result presented here is useful for such future studies.
Acknowledgements
The authors would like to thank Makoto Nishimoto, Masaki Yamamoto, and Tamami Ohara (National Institute of Technology (KOSEN), Wakayama College) for their assistance and invaluable advice during the course of this study.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
The full mitochondrial genome sequence reported in this study is registered in and openly available from the National Center for Biotechnology Information database (NCBI/GenBank) (Accession Number: LC600801; https://www.ncbi.nlm.nih.gov/nuccore/LC600801).
The specimen used in this study are vouchered at The University Museum of The University of Tokyo, Japan (Takenori Sasaki; [email protected]) under the voucher number UMUT RM33392.
Additional information
Funding
References
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. 1990. Basic local alignment search tool. J Mol Biol. 215:403–410.
- Bernt M, Donath A, Jühling F, Externbrink F, Florentz C, Fritzsch G, Pütz J, Middendorf M, Stadler PF. 2013. MITOS: improved de novo metazoan mitochondrial genome annotation. Mol Phylogenet Evol. 69(2):313–319.
- Castresana J. 2000. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol. 17(4):540–552.
- Cunha TJ, Giribet G. 2019. A congruent topology for deep gastropod relationships. Proc Royal Soc B. 286:20182776.
- Feng JT, Guo YH, Yan CR, Ye YY, Li JJ, Guo BY, Lü ZM. 2020. Comparative analysis of the complete mitochondrial genomes in two limpets from Lottiidae (Gastropoda: Patellogastropoda): rare irregular gene rearrangement within Gastropoda. Sci Rep. 10(1):1–4.
- Heath TA, Hedtke SM, Hillis DM. 2008. Taxon sampling and the accuracy of phylogenetic analyses. J Syst Evol. 46(3):239–257.
- Hillis DM, Pollock DD, McGuire JA, Zwickl DJ. 2003. Is sparse taxon sampling a problem for phylogenetic inference? Syst Biol. 52(1):124.
- Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 30(4):772–780.
- Nakano T, Ozawa T. 2007. Worldwide phylogeography of limpets of the order Patellogastropoda: molecular, morphological and palaeontological evidence. J Mollusc Stud. 73(1):79–99.
- Nakano T, Sasaki T. 2011. Recent advances in molecular phylogeny, systematics and evolution of patellogastropod limpets. J Mollusc Studies. 77(3):203–217.
- Nakano T, Sasaki T, Kase T. 2010. Colour polymorphism and historical biogeography in Japanese patellogastropod limpet Cellana nigrolineata (Reeve) (Patellogastropoda: Nacellidae). Zool Sci. 27(10):811–820.
- Pollock DD, Zwickl DJ, McGuire JA, Hillis DM. 2002. Increased taxon sampling is advantageous for phylogenetic inference. Syst Biol. 51(4):664.
- Ponder WF, Lindberg DR. 1997. Towards a phylogeny of gastropod molluscs: an analysis using morphological characters. Zool J Linnean Soc. 119(2):83–265.
- Powell AWB. 1973. The patellid limpets of the world (Patellidae). Indo-Pacific Mollusca. 3(15):75–206.
- Silvestro D, Michalak I. 2012. raxmlGUI: a graphical front-end for RAxML. Org Divers Evol. 12(4):335–337.
- Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 30(9):1312–1313.
- Stöger I, Schrödl M. 2013. Mitogenomics does not resolve deep molluscan relationships (yet?). Mol Phylogenet Evol. 69(2):376–392.
- Uribe JE, Irisarri I, Templado J, Zardoya R. 2019. New patellogastropod mitogenomes help counteracting long-branch attraction in the deep phylogeny of gastropod mollusks. Mol Phylogenet Evol. 1(133):12–23.
- Williams ST, Foster PG, Littlewood DT. 2014. The complete mitochondrial genome of a turbinid vetigastropod from MiSeq Illumina sequencing of genomic DNA and steps towards a resolved gastropod phylogeny. Gene. 533(1):38–47.
- Yang Z. 1994. Maximum likelihood phylogenetic estimation from DNA sequences with variable rates over sites: approximate methods. J Mol Evol. 39(3):306–314.