
Abstract
Sabia parviflora Wall. ex Roxb., an evergreen climbing woody vine, is a Chinese herbal medicine commonly used by ethnic minorities in some areas of China. In this study, the chloroplast genome of S. parviflora was sequenced for the first time. Its genome is 162,054 bp in length with 38.6% of GC content. The genome consists of a large single copy (LSC) region of 90,001 bp, a small single copy (SSC) region of 18,887 bp, and two inverted repeat (IRa and IRb) regions of 26,583 bp each. A total of 130 genes were annotated, including 85 protein-coding genes, 37 tRNA genes, and 8 rRNA genes. Phylogenetic analysis was conducted by nine species from order Proteales, which demonstrated a close relationship between the family Sabiaceae and Nelumbonaceae.
Sabia parviflora is an evergreen climbing woody vine of the genus Sabia (family Sabiaceae). It is native to Yunnan, Guizhou, and Guangxi province of China and is also distributed in India, Myanmar, Thailand, and other countries (ECFC Citation1988; Guo and Anthony Citation2007). As a traditional Chinese herbal medicine, S. parviflora is widely used to treat jaundice hepatitis, rheumatic arthritis, traumatic injury, and other diseases in some minority areas (Luo and Sun Citation2013). After recent years of research, it has been found that S. parviflora mainly contains terpenoids, alkaloids, flavonoids, etc., which have the effects of liver protection, antivirus, anti-rheumatic arthritis, anti-oxidation, and so on (Sun et al. Citation2019; Chen et al. Citation2020). At present, part of Chinese patent drugs and green tea have been developed with S. parviflora as raw material. However, as the most medicinally and economically valuable species in genus Sabia, the complete chloroplast genome of S. parviflora has not been reported, which restricted the taxonomic identification, phylogenetic analysis, and other studies of this genus. Therefore, this study sequenced the chloroplast genome of S. parviflora and the phylogenetic analysis was performed with species of Proteales.
Fresh leaves of S. parviflora were collected from Zhenning county, Guizhou province, China (E 105°36′24″, N 25°40′55″). Total DNA was extracted from those leaves using E.Z.N.A® Plant DNA Kit (OMEGA Bio-tek, Winooski, VT) according to the manufacturer’s instruction. The specimen and DNA sample were deposited in Herbarium of Guizhou University of Traditional Chinese Medicine (http://www.gzy.edu.cn/, Qingwen Sun, [email protected]) under the voucher number SABIA0728-07. The genome sequencing was conducted on the Illumina NovaSeq Sequencing System to generate paired-end 2*150 bp reads. Total about 5.43 Gb raw data were obtained. Trimmomatic (Bolger et al. Citation2014) was used to filter the raw data. NOVOPlasty (Dierckxsens et al. Citation2017) and CpGAVAS2 (Shi et al. Citation2019) were used to assemble and annotate the chloroplast genome, respectively, with the S. yunnanensis chloroplast genome sequences (NC_029431.1) as reference. The annotated genome sequence was submitted to the GenBank (Accession number: MW566751).
The chloroplast genome of S. parviflora is a double-stranded DNA molecule with a length of 162,054 bp. It presents a typical quadripartite structure, consisting of a large single copy (LSC) region, a small single copy (SSC) region, and two inverted repeat (IRa and IRb) regions. Its LSC and SSC regions are 90,001 and 18,887 bp, respectively, alternated by a pair of IR regions of 26,583 bp each. The overall GC content of this species is approximately 38.6% (LSC, 37.1%; SSC, 33.4%; IRs, 43.2%). A total of 130 genes were annotated, including 85 protein-coding genes, 37 tRNA genes, and 8 rRNA genes. Among these genes, six protein-coding genes (rps12, rps7, rpl23, rpl2, ndhB, and ycf2), seven tRNA genes (trnA-UGC, trnI-CAU, trnI-GAU, trnL-CAA, trnN-GUU, trnR-ACG, and trnV-GAC), and all rRNA genes (rrn16, rrn23, rrn4.5, and rrn5) including two repeating units. Furthermore, there are 19 genes contain one intron, including 11 protein-coding genes (atpF, ndhA, ndhB × 2, petB, petD, rpl16, rpl2 × 2, rpoC1, and rps16) and 8 tRNA genes (trnA-UGC × 2, trnG-UCC, trnI-GAU × 2, trnK-UUU, trnL-UAA, and trnV-UAC). A total of four genes contain two introns (rps12×2, clpP, and ycf3). In addition, two genes (ycf1 and rps19) were annotated as pseudogenes.
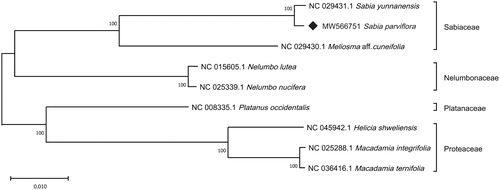
The whole chloroplast genomes of nine species in four families of Proteales were used for phylogenetic analysis. After sequence alignment by MAFFT (Katoh and Standley Citation2013), MEGA X (Kumar et al. Citation2018) was used to perform maximum likelihood (ML) tree with the Tamura Nei model. The bootstrap method was used to test the reliability of phylogeny with 1000 replicates. Two species from Sabia showed a cognate relationship, and the family Sabiaceae have a closer relationship with family Nelumbonaceae than that of Platanaceae and Proteaceae (). These relationships are congruent with previous report (Sun et al. Citation2016).
Figure 1. Phylogenetic relationships of nine Proteales species constructed from the complete chloroplast genome sequences using maximum likelihood (ML).
Disclosure statement
No potential conflict of interest was reported by the authors.
Data availability statement
The genome sequence data that support the findings of this study are openly available in GenBank of NCBI at [https://www.ncbi.nlm.nih.gov] (https://www.ncbi.nlm.nih.gov/) under the accession no. MW566751. The associated BioProject, SRA, and Bio-Sample numbers are PRJNA727714, SRR14455320, and SAMN19030660 respectively.
Additional information
Funding
References
- Bolger AM, Lohse M, Usadel B. 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 30(15):2114–2120.
- Chen YR, Pan GJ, Xu WF, Sun QW, Wang B, Zhang Y, Yang TJ. 2020. Spectrum-effect relationship study between HPLC fingerprints and antioxidant activity of Sabia parviflora. J Chromatogr B-Anal Technol Biomed Life Sci. 1140:121970.
- Dierckxsens N, Mardulyn P, Smits G. 2017. NOVOPlasty: de novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 45(4):e18.
- Editorial Committee of Flora of China (ECFC). 1988. Flora republicae popularis sinicae. Beijing: Science Press.
- Guo LX, Anthony RB. 2007. Flora of China (Sabiaceae) Vol. 12. Beijing: Science Press.
- Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 30(4):772–780.
- Kumar S, Stecher G, Li M, Knyaz C, Tamura K. 2018. MEGA X: molecular evolutionary genetics analysis across computing Platforms. Mol Biol Evol. 35(6):1547–1549.
- Luo YC, Sun QW. 2013. Natural medicines commonly used by ethnic groups in Guizhou. Vol. 2. Guiyang (China): Guizhou Science and Technology Publishing House.
- Shi LC, Chen HM, Jiang M, Wang LQ, Wu X, Huang LF, Liu C. 2019. CPGAVAS2, an integrated plastome sequence annotator and analyzer. Nucleic Acids Res. 47(W1):W65–W73.
- Sun QW, Pan GJ, Xu WF, Lu X, Bai CH, Liu MG, Chen YR. 2019. Isolation and structure elucidation of a new flavonol glycoside from Sabia Parviflora. Nat Prod Res. 29:1–6
- Sun YX, Moore MJ, Zhang SJ, Soltis PS, Soltis DE, Zhao TT, Meng AP, Li XD, Li JQ, Wang HC. 2016. Phylogenomic and structural analyses of 18 complete plastomes across nearly all families of early-diverging eudicots, including an angiosperm-wide analysis of IR gene content evolution. Mol Phylogenet Evol. 96:93–101.