
Abstract
Lindera praecox is a signature composition in the broadleaved deciduous forest of East China and Japan. Presently, the complete chloroplast (cp) genome of this species was sequenced, assembled, and annotated. It is 152,818 bp in length and encodes 85 protein-coding genes, 36 transfer RNA (tRNA) genes and eight ribosomal RNA (rRNA) genes. The phylogenetic analysis indicated intraspecific varieties within L. praecox species collected in China and Japan. This chloroplast genome sequencing offers genetic background for resources conservation and phylogenetic studies.
Lindera praecox (Siebold & Zuccarini) Blume is the member with extremely size of fruit in the genus Lindera, distributed in the broadleaved deciduous forest of Hubei, Anhui, Zhejiang and Japan, as a diagnostic component (Li et al. Citation2008). In present study, the completed chloroplast genome sequence of L. praecox is reported contributing for better understanding its evolution and population genetics, and providing significant information for the phylogeny of Lauraceae.
Genomic DNA was extracted from fresh leaves of a seedling of L. praecox from Bodaofeng, Luotian, Hubei, China (115.5865992°, 31.1391175°, 928 m, in the valley; Dong Hongjin et al. 1272, 2020-7-28; HIB, HTGC), the total genomic DNA was isolated according to a modified CTAB method (Doyle and Doyle Citation1987). Total genome DNA of L. praecox was sequenced by Illumina Hiseq 2500 Sequencing System (Illumina, Hayward, CA) to construct the shotgun library and assembled through the GetOrganelle software (Jin et al. Citation2020). The complete chloroplast genome of L. praecox was annotated with software PGA (Qu et al. Citation2019) and Geneious ver. 10.1 (Matthew et al. Citation2012) (http://www.geneious.com), and then submitted to GenBank (accession no. MW774641). The genome annotation was performed by aligning with the cp genomes of relatively related species.
The size of newly assembled chloroplast genome of L. praecox is 152,818 bp, including a large single-copy (LSC) region of 93,756 bp and a small single-copy (SSC) region of 18,910 bp separated by a pair identical inverted repeat (IR) region of 20,076 bp each. A total of 129 genes were successfully annotated containing 85 protein-coding genes, 36 tRNA genes and 8 rRNA genes. GC content of the whole genome, IRs, LSC and SSC regions are 39.1%, 44.4%, 37.9% and 33.9%, respectively. GC content of IRs region is the highest. 20 genes contain one intron, while 2 genes have two introns.
Compared to the chloroplast genome of L. praecox collected from Japan (Genbank accession no. MG581449), the China collection was 90 bp longer than Korea collection, 55 bp, 9 bp and 17 bp longer in LSC, SSC and IR regions, respectively. Totally, 220 variations were detected between the two plastomes, including 152 (69.1%) nucleotide substitutions and 68 indels. Among these mutations, 128 (58.2%) located in inter genic regions (IGRs), 88 (40%) in protein-coding regions (22 in intron) and four in tRNA genes’ intron. In summary, almost all of the mutations (66/68) in coding regions were substitution mutations, while 76/128 (59%) substitutions and 52/128 (41%) indels in IGS, 13/26 (50%) substitutions and indels respectively in intron. The comparison results indicated higher intra-species diversity such as Panax ginseng and Lonicera japonica (Kim et al. Citation2015; Kang et al. Citation2018).
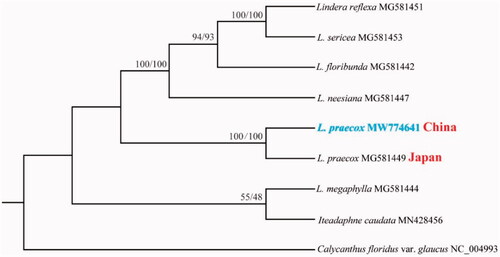
The complete chloroplast genome sequence of L. praecox and other species from Lauraceae were used to construct phylogenetic tree (). The sequences were initially aligned using MAFFT (Katoh and Standley Citation2013) and then visualized and manually adjusted using BioEdit (Hall Citation1999). Take the plastome of Calycanthus floridus var. glaucus (GenBank: NC_004993) as an out-group, a maximum likelihood analysis was performed with RAxML version 8 program (Alexandros Citation2014) using 1000 bootstrap. IQ-tree was also used to construct ML tree with fast mode (Nguyen et al. Citation2015). As expected, the result shows the chloroplast sequences of L. praecox from China and Japan were clustered together though the two sequences were different, and the position was consist with previous published topology (Jo et al. Citation2019). The results will be valuable for the genetic diversity study for this species.
Figure 1. Maximum likelihood phylogenetic tree for Lindera praecox based on complete chloroplast genomes. The number on each node indicates bootstrap support value generated by RaxML/IQtree.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
The data that support the findings of this study are available in GenBank of NCBI at https://www.ncbi.nlm.nih.gov, accession number MW774641. The assembled individual was linked with no. SAMN18324953 and Project ID: PRJNA715043.
Additional information
Funding
References
- Alexandros S. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 30:1312–1313.
- Doyle JJ, Doyle JL. 1987. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem Bull. 19:11–15.
- Hall TAB. 1999. A user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp Ser. 41:95–98.
- Jin JJ, Yu WB, Yang JB, Song Y, dePamphilis C, Yi TS, Li DZ. 2020. GetOrganelle: a fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 21(1):241.
- Jo S, Kim YK, Cheon SH, Fan Q, Kim KJ. 2019. Characterization of 20 complete plastomes from the tribe Laureae (Lauraceae) and distribution of small inversions. PLOS One. 14(11):e0224622.
- Kang SJ, Park HS, Koo HJ, Park JY, Lee D, Kang KB, Han S, Sung S, Yang TJ. 2018. The complete chloroplast genome sequence of Korean Lonicera japonica and intra-species diversity. Mitochondrial DNA Part B. 3(2):941–944.
- Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 30(4):772–780.
- Kim K, Lee SC, Lee J, Lee H, Joh H, Kim NH, Park HS, Yang TJ. 2015. Comprehensive survey of genetic diversity in chloroplast genomes and 45S nrDNAs within Panax ginseng species. PLOS One. 10(6):e0117159.
- Li XW, Li J, Huang PH, Wei FN, Van W. 2008. Lauraceae. Vol. 7. Beijing: Science Press.
- Matthew K, Richard M, Amy W, Steven SH, Matthew C, Shane S, Simon B, Alex C, Sidney M, Chris D. 2012. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 28:1647–1649.
- Nguyen LT, Schmidt HA, Arndt VH, Bui Quang M. 2015. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. 32(1):268–274.
- Qu XJ, Moore MJ, Li DZ, Yi TS. 2019. PGA: a software package for rapid, accurate, and flexible batch annotation of plastomes. Plant Methods. 15(1):1–12.