
Abstract
In this study, we sequenced, annotated, and analyzed the complete mitochondrial genome of Matsumuramata muiri (Kirkaldy, 1907) (Hemiptera: Delphacidae). The mitogenome was 16,276 bp in length with high A + T content (76.28%), containing 13 protein-coding genes, 22 tRNA genes, two rRNA genes, and a control region. Gene rearrangement of M. muiri was congruent with those of some reported delphacid species. All protein-coding genes initiated with ATN, except for nad5, which used non-canonical start codon GTG. The predicted secondary structures of all tRNA genes were typical cloverleaf except for trnS1 (AGN), lacking the dihydrouridine (DHU) stem.
Keywords:
Many species of Delphacidae (Hemiptera: Fulgoroidea) are economically significant pests of many important crops, such as rice and maize, because of serious crop yield losses caused by directly feeding, and by transmitting plant viral pathogens as disease vectors (Wilson Citation2005). Delphacidae is a diverse and cosmopolitan planthopper family with ∼2100 described species, of which most belong to the subfamily Delphacinae (Urban et al. Citation2010; Huang et al. Citation2017). Matsumuramata Xing & Chen, 2014, one group of Delphacinae, includes five species: M. muiri (Kirkaldy, 1907), M. sacchari (Matsumura, 1910), M. corporaali (Muir, 1923), M. parmenio (Fennah, 1969), and M. mani (Asche, 1988) (Bourgoin Citation2020). Little is known about the mitochondrial genomes of this genus. Here, we sequence and annotate the complete mitogenome of Matsumuramata muiri to facilitate a better understanding of the characters of delphacid mitogenomes and the evolutionary history of Delphacidae.
Adults of M. muiri were collected in Yingjiang County (N 24.71° and E 97.93°), Yunnan Province, China. The voucher specimen and its DNA were deposited at the Nanjing Institute of Environmental Sciences under the Ministry of Ecology and Environment, Nanjing, China (http://www.nies.org/, Yi Wu, [email protected]) under the voucher number (F4YN035). Total genomic DNA was extracted using the DNeasy Blood & Tissue Kit (Qiagen, Hilden, Germany) following the manufacturer’s protocols. A total of 15 primer pairs were chosen and modified from universal insect mitochondrial primers suggested in Simon et al. (Citation2006) to amplify the whole mitogenome. Purified target PCR products were sequenced directly on an ABI 3730XL DNA Analyzer using BigDye v3.1 (Applied Biosystems, USA). For those that sequence completely with difficulties, such as control region, PCR products were inserted into a pMD 19-T Vector (Takara Biomedical, Dalian, China), and multiple clones were sequenced independently. DNA sequences were edited and assembled into contigs by BioEdit 7.2.6.1 (Hall, 1999). After manually checking the assembly, the whole mitogenome was annotated with MitoZ v2.4-alpha (Meng et al. Citation2019). The annotation of tRNAs was verified by MITOS Web Server (Bernt et al. Citation2013). Start and stop codons of some PCGs were corrected according to the alignment of homologous genes in Fulgoroidea.
The complete mitochondrial of Matsumuramata muiri (Genbank accession no. MW288929) was 16,276 bp in size, with high A + T content (76.28%). The entire set of 37 genes was encoded, including 13 protein-coding genes (PCGs), 22 tRNA genes, two rRNA genes, and a control region, as observed in most insects. Gene rearrangement was detected to be congruent with those of other delphacid species, such as Sogatella furcifera, in which two gene clusters trnW-trnC-trnY and trnT-trnP-nad6 undertake conversion to trnC-trnW-trnY and nad6-trnP-trnT, respectively (Zhang et al. Citation2014).
The typical start codons ATN are used for twelve of all protein-coding genes (PCGs). The exception is nad5, which initiate with GTG. For all PCGs, three stop codons were used: T (atp6, cox1, and cox3), TAG (nad1 and nad3), and TAA (other eight PCGs). All 22 typical tRNA genes were found, ranging from 58 bp (trnS1 (AGN)) to 72 bp (trnK (CUU)). The predicted secondary structures of all tRNA genes are typical cloverleaf except for trnS1 (AGN), lacking the dihydrouridine (DHU) stem.
There were 14 overlaps in the mitogenome of M. muiri. The nad4l-nad4 overlap (ATGTTAA) was 7 bp in size, and not identical to that of atp8-atp6 (ATATTAA). A total of 13 non-coding regions were found, including 12 intergenic spacers, and the control region. The intergenic spacer between trnS2 (UCN) and nad1 was 16 bp in length, which corresponds to the binding sites of a transcription termination peptide. The control region spanned 1,768 bp locating between rrnS and trnI, and in this AT-rich region, a tandem unit (CATCGATTTTTGAAAAAAATG) was detected to repeat 25 times.
The amino acid sequence of each PCG was aligned individually using MAFFT v7.394 under the L-INS-i strategy (Katoh and Standley Citation2013). Then each alignment was trimmed and translated back into nucleotide sequence accordingly using trimAl v1.4.1. The nucleotide dataset of 13 PCGs were concatenated using FASconCAT-G v1.04 (Kück and Longo Citation2014). The optimal partitioning scheme and the best substitution model for each partition were selected using ModelFinder (Kalyaanamoorthy et al. Citation2017) implemented in IQ-TREE v1.6.10 (Nguyen et al. Citation2015). A maximum likelihood (ML) tree () was reconstructed with 1,000 replicates of ultrafast bootstrap in the IQ-TREE v1.6.10. The Bayesian inference () was performed using MrBayes 3.2.6 (Ronquist et al. Citation2012) with the best-fitting models found by ModelFinder. Philaenus spumarius (Hemiptera: Cercopoidea: Aphrophoridae) and one cixiid species Pentastiridius sp. (Hemiptera: Fulgoroidea: Cixiidae) were selected as outgroup.
Figure 1. Maximum likelihood (ML) and Bayesian Inference (BI) phylogenetic tree inferred from nucleotide sequences of 13 protein-coding genes. UFBoot support values (left) and Bayesian posterior probabilities (right) are indicated on nodes. Group names are marked by vertical lines. Matsumuramata muiri is marked with an asterisk.
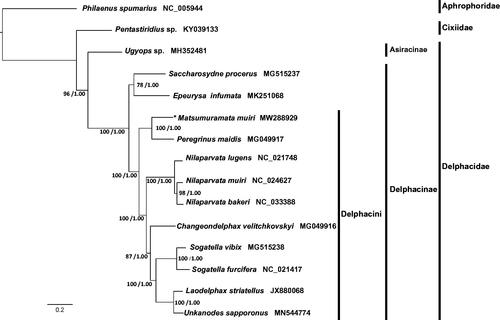
Cixiidae was a sister group to the family Delphacidae in the ML tree. Two clades were well-supported in Delphacidae, the subfamily Asiracinae clade represented by Ugyops sp. and the Delphacinae clade. Within Delphacini, Matsumuramata muiri and Peregrinus maidis were clustered together with strong support, indicating their relatively closed relationships. Nilaparvata lugens was sister to N. muiri + N. bakeri. The genus Sogatella seemed closed to both Unkanodes sapporonus and Laodelphax striatellus.
Disclosure statement
The authors report no conflicts of interest.
Data availability statement
The genome sequence data that support the findings of this study are openly available in GenBank of NCBI at https://www.ncbi.nlm.nih.gov/ under the accession no. MW288929.
References
- Bernt M, Donath A, Jühling F, Externbrink F, Florentz C, Fritzsch G, Pütz J, Middendorf M, Stadler PF. 2013. MITOS: improved de novo metazoan mitochondrial genome annotation. Mol Phylogenet Evol. 69(2):313–319.
- Bourgoin T. 2020. FLOW (Fulgoromorpha Lists on The Web): a world knowledge base dedicated to Fulgoromorpha. Version 8 [updated 2020 Nov 11]. http://hemiptera-databases.org/flow/ [accessed 2020 Nov 12].
- Huang YX, Zheng LF, Bartlett CR, Qin DZ. 2017. Resolving phylogenetic relationships of Delphacini and Tropidocephalini (Hemiptera: Delphacidae: Delphacinae) as inferred from four genetic loci. Sci Rep. 7(1):3319.
- Kalyaanamoorthy S, Minh BQ, Wong TKF, von Haeseler A, Jermiin LS. 2017. ModelFinder: fast model selection for accurate phylogenetic estimates. Nat Methods. 14(6):587–589.
- Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 30(4):772–780.
- Kück P, Longo GC. 2014. FASconCAT-G: extensive functions for multiple sequence alignment preparations concerning phylogenetic studies. Front Zool. 11(1):81.
- Meng G, Li Y, Yang C, Liu S. 2019. MitoZ: a toolkit for animal mitochondrial genome assembly, annotation and visualization. Nucleic Acids Res. 47(11):e63.
- Nguyen LT, Schmidt HA, von Haeseler A, Minh BQ. 2015. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. 32(1):268–274.
- Ronquist F, Teslenko M, van der Mark P, Ayres DL, Darling A, Höhna S, Larget B, Liu L, Suchard MA, Huelsenbeck JP. 2012. MRBAYES 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol. 61(3):539–542.
- Simon C, Buckley TR, Frati F, Stewart JB, Beckenbach AT. 2006. Incorporating molecular evolution into phylogenetic analysis and a new compilation of conserved polymerase chain reaction primers for animal mitochondrial DNA. Annu Rev Ecol Evol Syst. 37(1):545–579.
- Urban JM, Bartlett CR, Cryan JR. 2010. Evolution of Delphacidae (Hemiptera: Fulgoroidea): combined-evidence phylogenetics reveals importance of grass host shifts. Syst Entomol. 35(4):678–691.
- Wilson SW. 2005. Keys to the families of Fulgoromorpha with emphasis on planthoppers of potential economic importance in the southeastern United States (Hemiptera: Auchenorrhyncha). Florida Entomol. 88(4):464–481.2.0.CO;2]
- Zhang KJ, Zhu WC, Rong X, Liu J, Ding XL, Hong XY. 2014. The complete mitochondrial genome sequence of Sogatella furcifera (Horváth) and a comparative mitogenomic analysis of three predominant rice planthoppers. Gene. 533(1):100–109.