
Abstract
The Changbai Mountains comprise one of the main distribution areas of A. victorialis in China, and this species is endangered owing to habitat changes and overexploitation. However, A. victorialis germplasms have not been systematically collected and studied. The aims of this study were to obtain some detailed genetic information, analyze the genetic diversity, and further promote the protection of A. victorialis germplasms from the Changbai Mountains. Transcriptomic analysis was performed with six A. victorialis samples collected from the Changbai Mountains. At least 146,759 genes for each sample were obtained after performing de novo assembly of the RNA-seq data, and at least 92% of these genes were found to have only one mRNA isoform. These sequences and their functional annotations provided a large-scale genetic resource of this species. Phylogenetic analysis showed that A. victorialis was genetically distant from some related species, e.g. Allium sativum, Allium fistulosum, and Allium cepa, but genetically close to Allium tuberosum. The two A. victorialis var. listera samples were phylogenetically separated from the other four samples, and these two samples should be regarded as Allium listera. In addition, two KASP markers for discriminating the Dongfeng samples from the other four A. victorialis samples were successfully developed. This study lays the foundation for future studies on the genetic diversity and evolution of Allium species, as well as for the conservation of A. victorialis germplasms from the Changbai Mountains and other populations of this species.
1. Introduction
Allium victorialis, a perennial herb in the section Anguinum of the genus Allium, is widely distributed in the northern hemisphere. In China, it is mainly distributed in Northeast China, Inner Mongolia, and Hebei Province. Allium victorialis has great economic potential, with a long history of usage as both a functional food and a traditional medicine. Recently, this species has been of increasing interest, particularly in regard to its nutritional quality and active ingredients (Lee et al. Citation2001; Khan et al. Citation2013; Woo and Lee Citation2013). Moreover, A. victorialis has a wide geographical distribution and a relatively old evolutionary geographical history in the section Anguinum and therefore can be considered a primitive species in the section Anguinum, making it crucial to genetic and evolutionary research of Allium species (Jing et al. Citation1999). However, there are many subspecies and varieties of A. victorialis (Yang et al. Citation2014; Herden et al. Citation2016), which hinders the genetic studies and further utilization of A. victorialis. Therefore, more research is needed to further elucidate the genetics, evolution, and taxonomy of this species. To date, few studies have explored the genetic diversity of A. victorialis. Previous studies focused mainly on the analysis of the intraspecific genetic relationships of A. victorialis or the genetic relationships between A. victorialis and other Allium species based on external morphology (Yoo et al. Citation1998), karyotype analysis (Jing et al. Citation1999), random amplified polymorphic DNA (RAPD) molecular markers (Lim et al. Citation1998), sequencing of the internal transcribed spacer (ITS) region from the nuclear ribosomal DNA (Herden et al. Citation2016), and sequencing of the chloroplast DNA (Li et al. Citation2010; Herden et al. Citation2016). However, these previous studies could not fully explain the intraspecific genetic diversity of A. victorialis or the genetic diversity between A. victorialis and other Allium species.
As eukaryotes, many Allium species have large and complex genome sizes of 10–20 Gbp, which leads to difficulties in genetic research of Allium species (Kamenetsky et al. Citation2015; Barboza et al. Citation2018). Recently, next-generation sequencing (NGS) technology has been widely used as an important tool in the research of plant genetics. Unlike genome analysis, transcriptome analysis by NGS technology is rapid, inexpensive, and unconstrained by genomic complexity (Zhu et al. Citation2017). Additionally, this technique has been effectively applied to several Allium species, including onion (Rajkumar et al. Citation2015; Han et al. Citation2016), garlic (Sun et al. Citation2012; Chen et al. Citation2018), and Chinese chive (Zhou et al. Citation2015). Transcriptome sequencing can provide a wealth of biological information and is an effective method for gene functional annotation (Sun et al. Citation2012), gene expression analysis (Khosa et al. Citation2016), molecular marker development (Liu et al. Citation2015), and interspecific genetic diversity analysis (Zhu et al. Citation2017). However, studies that apply transcriptome analysis to improve the understanding of the interspecific genetic relationships of A. victorialis are still lacking.
Recently, researchers have attempted to understand the genetic structures and variations in plant genomes. Various molecular markers, such as amplified fragment length polymorphism (AFLP), simple sequence repeats (SSRs), and single nucleotide polymorphisms (SNPs), have been applied to investigate the genetic diversity of plant species (Li et al. Citation2019). SNPs have become the preferred markers for numerous genetics and genomics applications (Li et al. Citation2019). In particular, the revolutionary advances in genome sequencing (Ryu et al. Citation2018; Han et al. Citation2020) have made the SNP-based genotyping technology an effective method for large-scale genotyping, genetic diversity analyses, and genetic map construction in many plant species (Li et al. Citation2019). Kompetitive allele-specific PCR (KASP) is a novel and extensively used genotyping technology that enables bi-allelic scoring of SNPs (Han et al. Citation2019). This method has been successfully applied in the genotyping and identification of different varieties or materials within species or among related species (Fang et al. Citation2020; Ma et al. Citation2021). However, this technology has still not been extensively applied in Allium species, especially in A. victorialis.
The Changbai Mountains in Jilin Province comprise one of the main distribution areas of A. victorialis in Northeast China, where this is an important species. In recent years, considerable habitat changes and human overexploitation have posed serious threats to A. victorialis germplasms from this region, resulting in a population decline and endangerment of this species. Thus, the Changbai Mountain populations of this species are in urgent need of effective conservation. However, A. victorialis germplasms from this region have not been systematically collected and studied; therefore, their current population dynamics and germplasms are unclear. The purposes of this study were to obtain detailed genetic information and to analyze the genetic diversity of A. victorialis germplasms from the Changbai Mountains using transcriptomic methods, as well as to develop SNP markers for discriminating A. victorialis germplasms from the Changbai Mountains and those of other Allium species. The results of this study would not only aid in enacting conservation policies for A. victorialis germplasms from this region but also provide a reference for the study of other populations of this species.
2. Materials and methods
2.1. Plant material
Six A. victorialis samples were collected from the Changbai Mountains, Jilin Province, China. Two samples were collected from Dunhua (43°10′27″ N, 128°02′17″ E), one from Wangqing (43°50′49″ N, 130°10′36″ E), one from Antu (42°58′11″ N, 128°33′57″ E), and two from Dongfeng (43°04′24″ N, 125°32′58″ E). All samples were deposited in the herbarium of the Jilin Provincial Academy of Forestry Sciences and identified with reference to Tang and Wang (Citation1980). The two Dongfeng samples were identified as Allium victorialis var. listera, while the other four were identified as A. victorialis (, ).
Figure 1. Six Allium victorialis samples from the Changbai Mountains, Jilin Province, China. (A) Dongfeng1, (B) Dongfeng2, (C) Dunhua1, (D) Dunhua2, (E) Antu, and (F) Wangqing.

Table 1. Leaf morphology of six Allium victorialis samples.
Eight to ten healthy plants were collected for each sample during the leaf development period and cleaned with water. Then, leaf length and width were measured to calculate the length-to-width ratio as a leaf index. The leaf length, leaf width, and leaf index were measured in three replications. Next, the plants were immediately frozen in liquid nitrogen in the laboratory, and the healthy leaf tissues of each plant were stored at −80 °C until RNA extraction.
2.2. RNA extraction, library construction, and transcriptome analysis
RNA was extracted from the leaf tissues using an RNA Easy Fast Plant Tissue Kit (TIANGEN Biotech, Beijing, China). After the RNA samples passed the RNA purity and integrity tests, they were randomly fragmented and then reverse-transcribed into cDNA using a random hexamer primer, followed by end repairing, polyA tailing, sequencing adapter ligation, and PCR amplification to construct a transcriptome library. The constructed transcriptome library was sequenced on an Illumina NovaSeq 6000 platform at Novogene (Beijing, China). Low-quality reads and adapters were removed from the original sequencing data to obtain clean reads, which were de novo assembled using rnaSPAdes v3.13.1 (Bushmanova et al. Citation2019). As full-length mRNA sequences may not be generated using the de novo assembly method, each scaffold was considered a transcript fragment, and transcripts with the same gene number were identified from the rnaSPAdes output FASTA file. Then, the transcript number and the longest transcript for each gene were determined. Transcriptome assembly a completeness was assessed by benchmarking universal single-copy ortholog (BUSCO) analysis with the Eukaryota sets of BUSCO v3 (Waterhouse et al. Citation2018).
2.3. Open reading frame (ORF) prediction and protein domain annotation
For each sample, the ORFs of each assembled transcript were predicted using the script transcript_fasta_to_ORF_pics.pl in Trinity v2.8.5 (Grabherr et al. Citation2011), and the longest ORF was selected to represent the transcript. In addition, as genes may have multiple isoforms, we also selected the ORF of the longest isoform of each gene for the gene-level analysis. Protein domains were searched against the Pfam v31.0 database based on the ORF-encoded amino acid sequences using HMMER v3.1b1 (Finn et al. Citation2016).
2.4. Phylogenetic tree construction
The ORFs of A. victorialis and other species were subjected to gene family analysis using OrthoFinder (Emms and Kelly Citation2019). Based on gene family analysis, Asparagus officinalis and Arabidopsis thaliana were used as the outgroups, and a total of 103 single-copy genes that were present in the six samples, related Allium species (A. cepa, A. fistulosum, A. ascalonicum, A. macrostemon, A. ampeloprasum, A. chinense, A. sativum, and A. tuberosum), and the outgroups were selected to construct the phylogenetic tree (Harkess et al. Citation2017; Zhu et al. Citation2017; Chen et al. Citation2018; Emms and Kelly Citation2019; http://www.arabidopsis.org/).
The selected single-copy gene sequences were aligned using MUSCLE v3.8.31 (Edgar Citation2004) and then concatenated to perform ProTest v3.4 (Darriba et al. Citation2011) analysis. This analysis determined the optimal model for phylogenetic tree construction, i.e. JIT + F+G + I, namely the Jones–Taylor–Thornton (JTT; Jones et al. Citation1992) model incorporating empirical frequencies (+F) and invariant sites (+I) along with discrete gamma rate categories (+G), according to two model selection criteria: Akaike information criterion (Akaike Citation1973) and Bayesian information criterion (Schwarz Citation1978). A maximum-likelihood phylogenetic tree with 500 bootstrap replicates was constructed using Mega v7 (Kumar et al. Citation2018) and RAxML (Stamatakis Citation2014), with both presenting similar results.
2.5. Development and validation of KASP markers
The SNPs detected from sequence alignments of 103 single-copy genes from A. victorialis germplasms and other eight Allium species were used to develop KASP markers. A total of 14 SNPs, which satisfied the requirement of no other polymorphic site within 30-bp regions away from the SNPs, were used to design KASP markers. Sequences of 200 bp upstream and downstream of the distinctive SNPs were extracted and used for primer design using both DNAMAN v6.0 (https://www.lynnon.com/dnaman.html) and Primer3Plus v2.4.2 (http://www.primer3plus.com/). PCR products were 70–120 bp long.
Primer Mix was set up as recommended by the Laboratory of the Government Chemist (LGC, http://www.lgcgenomics.com/), from which the KASP Master Mix Kit was purchased. The assay was tested in a 384-well format and set up as a 5-μL reaction containing 37.5 ng of template DNA (2.5 μL 15 ng/μL template DNA), 2.5 μL 2× KASP Master Mix, and 0.07 μL specific Primer Mix. PCR was performed under the following conditions: the template was denatured at 94 °C for 15 min, with 10 cycles of denaturation, annealing, and elongation (94 °C for 20 s, 61–55 °C for 60 s; decreasing by 0.6 °C per cycle), followed by 32 cycles of denaturation, annealing, and elongation (94 °C for 20 s and 55 °C for 60 s). Fluorescence was detected using a 7900HT Fast Real-Time PCR System (Applied Biosystems, Foster City, CA). Data analysis was performed manually using the inbuilt SDS v2.3 software (option of Allelic Discrimination).
3. Results
3.1. Morphological characteristics of A. victorialis
The morphological characteristics such as the bulbs, pedicels, perianths, filaments, and ovaries were all similar among the six samples. The bulbs of these six samples were all cylindrical to conical, solitary, or clustered. The pedicels of these six samples were almost identical and 2–3 times longer than the perianth. The perianths were yellowish-white, and some perianths of the two Dunhua samples were tinged with purple. The outer perianths of these six samples were boat-shaped, whereas the inner perianths were elliptic to ovate. The filaments of these six samples were from 0.25 to 1 times longer than the perianth, connate at the base, and adnate to perianth segments. The ovaries of these six samples were all constricted at the base into a short stipe and had three round edges, with one ovule per locule. However, the morphological differences in leaves among the six samples were more apparent. The leaf length of Dongfeng and Dunhua samples was 2–3 times their leaf width, with leaf indices of 2.35 (Dongfeng1), 2.24 (Dongfeng2), 2.57 (Dunhua1), and 2.84 (Dunhua2; ). The leaves of the two Dongfeng samples were elliptic to ovate and bases were cordate to rounded, while the leaves of the two Dunhua samples were oblanceolate-elliptic to elliptic and bases were cuneate to broad, with curling and folds. Furthermore, the leaf length was different from the leaf width in the Wangqing sample (leaf index, 3.47), and this difference was especially evident in the Antu sample, wherein the leaves were relatively long and narrow with a leaf index of 5.27, the leaves of these two samples were all oblanceolate-elliptic to elliptic, and bases were cuneate to broad ().
3.2. Transcript assembly
A total of 752.37 × 106 raw reads were generated from the six A. victorialis samples. The raw sequencing data used in the study are available at the NCBI SRA database with the accession numbers SRR11818586–SRR11818591. After filtering adapters and low-quality reads, 730.63 × 106 clean reads of 1095.94 × 108 bases remained, accounting for 97.11% of total raw reads (Table S1).
Based on these clean reads, de novo assembly was performed to obtain the transcripts for each sample. Among the six samples, transcript fragments that ranged from 159,228 (belonging to 146,759 gene fragments; Antu) to 314,196 (belonging to 291,564 gene fragments; Dunhua2) were obtained (Table S2). At least 92.86% of the gene fragments had only one isoform (Table S2). The longest transcripts in these samples ranged from 10,496 (Dongfeng1) to 15,950 bp (Dunhua1) (Table S3). The number of transcripts with a length of more than 1000 bp ranged from 28,684 (Antu) to 53,702 (Wangqing) (Table S3). The large number of long transcripts suggested that the analysis comprised a large number of full-length mRNAs. Moreover, the N50 values of the assemblies were 738–1429 bp, and the GC contents were 36.7–38.7% (Table S3).
We also assessed the quality of the assemblies by performing a BUSCO assessment. As shown in Table S4, the complete BUSCO ratios of the gene fragments and transcript fragments of A. victorialis were all higher than 84.8%, which suggested that high-quality cDNA sequences of this species were identified.
3.3. ORF prediction and Pfam-based domain annotation
For the six samples, ORFs of all transcript fragments were annotated, and as genes may have multiple isoforms, the ORF of the longest isoform of each gene was selected for the gene-level analysis. In each sample, at least 64% of transcripts and 61% of genes had ORFs (Table S5). The transcript-level ORF numbers ranged from 115,540 (Antu) to 201,996 (Dunhua2), and the gene-level ORF numbers ranged from 103,426 (Antu) to 180,504 (Dunhua2) (Table S5).
Based on the ORF sequences, the functional domains were annotated. The analysis showed that 28,184 (Antu) to 47,604 (Dunhua1) transcripts and 22,419 (Antu) to 29,731 (Dunhua1) genes had functional domains, accounting for less than 16.15% of the genes and 18.67% of the transcripts (Table S5). This result indicates that the functions of many important genes (or transcripts) from A. victorialis remain to be clarified.
We also compared the domain annotations of six A. victorialis samples with that of the other eight Allium species, including A. tuberosum, A. sativum, A. ampeloprasum, A. macrostemon, A. chinense, A. ascalonicum, A. cepa, and A. fistulosum (Zhu et al. Citation2017; Chen et al. Citation2018). A total of 3765 Pfam families were conserved among the nine species. Among these families, the protein kinase domain and protein tyrosine kinase domain were most prominently represented in the Pfam database. In addition, the protein domains that were also well-represented in A. victorialis were those for reverse transcriptase (RNA-dependent DNA polymerase), PPR repeat family, PPR repeat, RNA recognition motif (a.k.a. RRM, RBD, or RNP domain), and cytochrome P450 (Table S6).
3.4. Phylogenetic relationships of Allium species
Phylogenetic analysis revealed that the six A. victorialis samples were grouped into one clade with A. tuberosum, with a relatively close genetic relationship (bootstrap value, BS = 99), but they were on different branches (). The related Allium species, A. sativum, A. ampeloprasum, A. macrostemon, A. chinense, A. ascalonicum, A. cepa, and A. fistulosum, were phylogenetically distant from A. victorialis based on their position in a different clade.
Figure 2. Phylogenetic tree of Allium victorialis and other Allium species. The value above each clade is the bootstrap value (BS) of the maximum-likelihood estimation. The gene sequence information of other Allium species was adapted from Harkess et al. (Citation2017), Zhu et al. (Citation2017), Chen et al. (Citation2018), and TAIR10 (https://www.arabidopsis.org/).
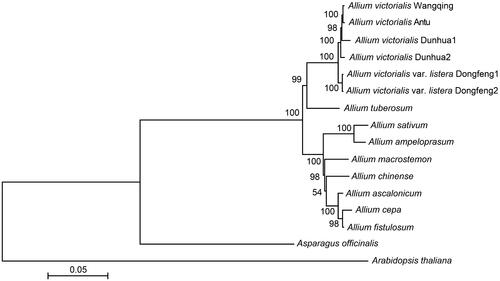
Among the six A. victorialis samples, the two Dongfeng samples were grouped into one sub-clade, while the other four samples were grouped into another sub-clade. The four samples were further clustered into three clades, namely, two separate sub-clades containing Dunhua1 and Dunhua2 (, BS = 100), and one other sub-clade containing both Antu and Wangqing (, BS = 100). Despite the close genetic relationship among A. victorialis samples from the Changbai Mountains, the results also indicated that there was a certain genetic distance among these A. victorialis samples, indicating a certain level of genetic diversity among them.
3.5. Development of KASP markers for A. victorialis
A total of 14 KASP markers were developed to further identify the relationships among A. victorialis germplasms. Ten of the 14 markers successfully distinguished the six A. victorialis samples and the other eight Allium species selected in this study (Table S7). As shown in Figures S1–S10, the A_SNP5A1 and A_SNP6A1 markers were successfully used to differentiate between the two Dongfeng samples and the other four A. victorialis samples from the Changbai Mountains, and the two Dongfeng samples were more related with the other Allium species analyzed in this study. The A_SNP8A1 and A_SNP9A1 markers were used to distinguish A. victorialis samples and some other Allium species used in this study (excluding A. tuberosum). However, the other markers produced unexpected results in distinguishing the six A. victorialis samples and the other Allium species in this study, suggesting that these polymorphic nucleotides identified from RNA-seq might not be species-specific. These results showed that KASP technology can be used for distinguishing A. victorialis germplasms from the Changbai Mountains and other Allium species.
4. Discussion
4.1. Transcriptome analysis
It is a challenge to investigate the genetics of Allium species because most Allium species have large and complex genome sizes of 10–20 Gbp (Kamenetsky et al. Citation2015; Barboza et al. Citation2018). Recently, NGS technology has been applied to the genetic research of Allium species as a strategy to effectively obtain the whole-genome mRNA data, although a large number of incomplete or redundant transcripts in Allium species may exist (Zhu et al. Citation2019). In our study, the annotation rates of the obtained transcriptome were 15.30–18.67% in the six A. victorialis samples. However, the annotation rates were higher than 20% in the Pfam database for some other Allium species, such as onion (≥21.0%) (Zheng et al. Citation2016; Zhu et al. Citation2017; Zhang et al. Citation2018) and garlic (≥24.9%) (Liu et al. Citation2015; Zhu et al. Citation2017). In addition, there were some protein domains in the Pfam database that were conserved in A. victorialis and the other related Allium species used in this study, such as the protein kinase domain and protein tyrosine kinase domain. This might be due to the difficulties in whole-genome transcriptome assembly, considering the large genome size of Allium species.
4.2. Phylogenetic tree-based analysis of genetic relationship and diversity of A. victorialis germplasms
Although previous molecular genetics studies have investigated the intraspecific genetic diversity of A. victorialis or interspecific genetic diversity between A. victorialis and other Allium species, the focus of these studies was mainly on A. victorialis in regions such as Japan, Korea, Europe, and North America (Lim et al. Citation1998; Inatomi et al. Citation2004; Herden et al. Citation2016). Thus far, studies on the molecular genetics of A. victorialis from the Changbai Mountains have not yet been reported. Furthermore, there are many subspecies and varieties of A. victorialis (Yang et al. Citation2014; Herden et al. Citation2016), leading to some difficulties in the identification of A. victorialis germplasms. For example, the two Dongfeng samples were identified as A. victorialis var. listera based on the similarity of their floral organ morphology to that of A. victorialis, but their leaf shape was different from the leaf shape of A. victorialis. Therefore, no consensus has been reached on the identification of A. victorialis var. listera. Some morphological studies have proposed that A. victorialis var. listera should be regarded as a variety of A. victorialis (Tolgor Citation1991; Tolgor et al. Citation1993), but some other studies based on karyotype analysis (Jing et al. Citation1999) and molecular systematics (Li et al. Citation2010) have suggested that this taxon should be renamed as Allium listera, as a separate species closely related to A. victorialis. To our knowledge, the present study is the first to systematically analyze the genetic diversity of A. victorialis germplasms from the Changbai Mountains, in particular, the diversity between the Dongfeng samples and those from other habitats in this region, by constructing a phylogenetic tree derived from transcriptome analysis. The two A. victorialis var. listera samples from Dongfeng were clustered together, whereas the other four samples were clustered separately, indicating a certain genetic distance between the so-called A. victorialis var. listera and A. victorialis. Thus, A. victorialis var. listera should be regarded as A. listera, consistent with some previously reported findings (Jing et al. Citation1999; Li et al. Citation2010). This divergence may be because A. victorialis, as a species in the section Anguinum, differentiated early with a high phylogenetic position, and there are three types (diploid, triploid, and tetraploid) in this species. The original population of A. victorialis is diploid (Jing et al. Citation1999; Li et al. Citation2010). Allium listera is in the section Anguinum and is endemic to China. Allium listera has the same origin as A. victorialis but is diploid only. The distribution area of A. listera is at the outer edge of the distribution area of the diploid A. victorialis and is homologous to the tetraploid A. victorialis (Herden et al. Citation2016; Lu et al. Citation2017).
In the Changbai Mountains, A. listera is mainly distributed in Hancongding in Dongfeng County, which is at the southern edge of the distribution area of A. victorialis in Jilin Province. Similar to the tetraploid A. victorialis in Dunhua, A. listera derived from the original diploid A. victorialis. During evolution, some diploid A. victorialis populations underwent polyploidization and asexual reproduction. Then, these diploid populations finally evolved into the tetraploid populations, probably due to an enhanced adaption to more stressful environmental conditions (Jing et al. Citation1999; Lu et al. Citation2017). In contrast, some diploid plants did not change their number of chromosomes over their long-term evolutionary process and maintained their own genetic characteristics in reproductive isolation from the tetraploid A. victorialis, evolving independently into A. listera, which has a wider ecological amplitude.
Among the other four A. victorialis samples, Dunhua1 and Dunhua2 were clustered separately, and their leaf morphologies were similar, with a leaf index of 2.0–3.0. The sample Dunhua2 was introduced from the more northern Heilongjiang Province and had wide leaves, whereas the sample Dunhua1 was collected from the Changbai Mountains and had wide leaves with a large plant type. However, the other two samples (from Wangqing and Antu) were clustered together. They were both A. victorialis germplasms from the Changbai Mountains, bearing narrow lanceolate leaves with a large leaf index. The habitat of the Wangqing sample was at higher latitudes and in greater proximity to Heilongjiang Province. Therefore, the Wangqing sample may represent a transitional type of A. victorialis from the Changbai Mountains to Heilongjiang Province, whereas the habitat of the Antu sample was closer to the traditional distribution area of A. victorialis in the Changbai Mountains. This demonstrates that the genetic diversity, origin, and habitat of A. victorialis affected its growth, reflecting a correlation between the growth conditions, genetic background, and habitats of this species.
Among the other reported Allium species, we found that the genetic relationship between A. victorialis and A. tuberosum was relatively close, but they were genetically distant from the other related Allium species discussed in the present study. This corroborates some previous studies on the genetic relationship between A. victorialis and other Allium species by DNA sequencing (Friesen et al. Citation2006; Li et al. Citation2010; Abugalieva et al. Citation2017). Allium victorialis belongs to the section Anguinum, a monophyletic group with an ancient origin and a wide distribution (Jing et al. Citation1999; Li et al. Citation2010), which evolved along the second evolutionary line of the three evolutionary lines of the genus Allium (Friesen et al. Citation2006; Li et al. Citation2010). In contrast, A. tuberosum, A. macrostemon, A. sativum, A. ampeloprasum, A. chinense, A. fistulosum, and A. cepa separately belong to one of the four Allium sections, namely Butomissa, Sacculiferum, Allium, and Cepa, all of which have certain genetic distances from the section Anguinum. The distance between the sections Anguinum and Butomissa was smaller than the distance between the section Anguinum and the other three sections, which suggests that the sections Butomissa and Anguinum may have a common ancestor (Friesen et al. Citation2006; Li et al. Citation2010; Abugalieva et al. Citation2017). All the above seven species belong to the third evolutionary line of the genus Allium, and their evolutionary sequence was more recent than the sequence of the section Anguinum (Friesen et al. Citation2006; Li et al. Citation2010).
Regarding morphological and growth characteristics, the seeds of A. victorialis have hypogeal germination and epicotyl dormancy (Kawano and Nagai Citation2005; Friesen et al. Citation2006). This species has reticulate-fibrous bulb tunics, prominent rhizomes, and narrow, branched, and lengthwise twisted septal nectaries (Friesen et al. Citation2006), with many of these characteristics being unique to the section Anguinum. However, the leaves of A. sativum and A. tuberosum are flat and solid; the leaves of A. fistulosum, A. ascalonicum, A. cepa, A. macrostemon, A. ampeloprasum, and A. chinense are hollow and fistular; and A. sativum, A. cepa, A. macrostemon, and A. ampeloprasum have swollen bulbs with membranous, papery, or leathery tunics, all of which are distinctly different from those of A. victorialis, indicating that the species evolved along different evolutionary lines.
4.3. Identification of A. victorialis germplasms by KASP assays
KASP technology is an effective and widely used genotyping method that determines the alleles at a specific locus within genomic DNA (Han et al. Citation2019; Li et al. Citation2019). This method has facilitated plant genetic diversity studies, for example, in the genotyping of Rubus (Ryu et al. Citation2018), non-heading Chinese cabbage (Li et al. Citation2019), and persimmon (Ma et al. Citation2021). However, it has not been previously applied in the analysis of A. victorialis germplasms. In this study, a KASP assay was used to further distinguish A. victorialis samples from the Changbai Mountains and some other Allium species. Four KASP markers failed to show polymorphisms among different Allium species. This was probably due to the relatively large differences between A. victorialis and the other Allium species, the presence of duplicated loci, unsuccessful primer design near the SNP regions, or the need to optimize PCR conditions (Ryu et al. Citation2018; Ma et al. Citation2021). Two successful markers (A_SNP5A1 and A_SNP6A1) could be used to discriminate the two Dongfeng samples and the other four A. victorialis samples, although they did not adequately distinguish genetical differences among the samples from Dunhua, Antu, and Wangqing. The KASP assay results indicated that the Dongfeng samples were clearly separated from the other four samples from the Changbai Mountains, which is consistent with the results based on the phylogenetic tree analysis. To our knowledge, this is the first report on the development of KASP markers in A. victorialis. With the emerging advancements in genomic technologies and assembly methods, the sequences of the very large A. victorialis genomes can be obtained in future studies, which would benefit the development of KASP markers to effectively discriminate A. victorialis germplasms.
5. Conclusions
A transcriptomic analysis of six different A. victorialis samples from the Changbai Mountains, China, was performed. Based on transcriptome libraries, we constructed a phylogenetic tree containing A. victorialis and other related Allium species, such as A. tuberosum, A. sativum, A. fistulosum, and A. cepa. The six A. victorialis samples were clustered into two clades – one containing the two Dongfeng samples and the other containing the other four samples. Allium victorialis exhibited certain genetic distances from the related Allium species and was relatively close to A. tuberosum, which suggests that they may have a common ancestor. Furthermore, Allium species could be distinguished using KASP markers, including two KASP markers that could distinguish the Dongfeng samples and the other four A. victorialis samples. This is the first study to apply transcriptome analysis to explore the genetic diversity of A. victorialis. Therefore, these findings not only provide a large-scale genetic resource for future genetic diversity and evolutionary research on A. victorialis from the Changbai Mountains but also lay the foundation for the conservation of this species.
Supplemental Material
Download Zip (9 MB)Disclosure statement
No potential conflict of interest was reported by the authors.
Data availability statement
The data that support the findings of this study are openly available in figshare at http://dx.doi.org/10.6084/m9.figshare.12769778. The sequencing reads supporting our findings are openly available in the NCBI SRA database (https://www.ncbi.nlm.nih.gov; accession numbers: SRR11818586–SRR11818591).
Additional information
Funding
Related Research Data
References
- Abugalieva S, Volkova L, Genievskaya Y, Ivaschenko A, Kotukhov Y, Sakauova G, Turuspekov Y. 2017. Taxonomic assessment of Allium species from Kazakhstan based on ITS and matK markers. BMC Plant Biol. 17(Suppl. 2):258.
- Akaike H. 1973. Information theory and an extension of the maximum likelihood principle. In: Petrov BN, Csaki F, editors. Proceedings of the 2nd International Symposium on Information Theory. Budapest, Hungary: Akadémiai Kiadó. p. 267–281.
- Barboza K, Beretta V, Kozub PC, Salinas C, Morgenfeld MM, Galmarini CR, Cavagnaro PF. 2018. Microsatellite analysis and marker development in garlic: distribution in EST sequence, genetic diversity analysis, and marker transferability across Alliaceae. Mol Genet Genomics. 293(5):1091–1106.
- Bushmanova E, Antipov D, Lapidus A, Prjibelski AD. 2019. rnaSPAdes: a de novo transcriptome assembler and its application to RNA-Seq data. GigaScience. 8:1–13.
- Chen X, Liu X, Zhu S, Tang S, Mei S, Chen J, Li S, Liu M, Gu Y, Dai Q, et al. 2018. Transcriptome-referenced association study of clove shape traits in garlic. DNA Res. 25(6):587–596.
- Darriba D, Taboada GL, Doallo R, Posada D. 2011. ProtTest 3: fast selection of best-fit models of protein evolution. Bioinformatics. 27(8):1164–1165.
- Edgar RC. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32(5):1792–1797.
- Emms DM, Kelly S. 2019. OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biol. 20(1):238.
- Fang T, Lei L, Li G, Powers C, Hunger RM, Carver BF, Yan L. 2020. Development and deployment of KASP markers for multiple alleles of Lr34 in wheat. Theor Appl Genet. 133(7):2183–2195.
- Finn RD, Coggill P, Eberhardt RY, Eddy SR, Mistry J, Mitchell AL, Potter SC, Punta M, Qureshi M, Sangrador-Vegas A, et al. 2016. The Pfam protein families database: towards a more sustainable future. Nucleic Acids Res. 44(D1):D279–D285.
- Friesen N, Fritsch R, Blattner F. 2006. Phylogeny and new intrageneric classification of Allium (Alliaceae) based on nuclear ribosomal DNA ITS sequences. Aliso. 22(1):372–395.
- Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, Adiconis X, Fan L, Raychowdhury R, Zeng Q, et al. 2011. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol. 29(7):644–652.
- Han F, Zhang X, Yuan K, Fang Z, Yang L, Zhuang M, Zhang Y, Wang Y, Liu Y, Li Z, et al. 2019. A user-friendly KASP molecular marker developed for the DGMS-based breeding system in Brassica oleracea species. Mol Breed. 39(6):90.
- Han G, Liu S, Jin Y, Jia M, Ma P, Liu H, Wang J, An D. 2020. Scale development and utilization of universal PCR-based and high-throughput KASP markers specific for chromosome arms of rye (Secale cereale L.). BMC Genomics. 21(1):206.
- Han J, Thamilarasan SK, Natarajan S, Park JI, Chung MY, Nou IS. 2016. De novo assembly and transcriptome analysis of bulb onion (Allium cepa L.) during cold acclimation using contrasting genotypes. PLoS One. 11(9):e0161987.
- Harkess A, Zhou J, Xu C, Bowers JE, Van der Hulst R, Ayyampalayam S, Mercati F, Riccardi P, McKain MR, Kakrana A, et al. 2017. The asparagus genome sheds light on the origin and evolution of a young Y chromosome. Nat Commun. 8(1):1279.
- Herden T, Hanelt P, Friesen N. 2016. Phylogeny of Allium L. subgenus Anguinum (G. Don. ex W.D.J. Koch) N. Friesen (Amaryllidaceae). Mol Phylogenet Evol. 95:79–93.
- Inatomi Y, Murata N, Nakano H, Tamura H, Suzuki T, Oosawa K. 2004. Variations on sprouting time and classification by RAPD analysis of Allium victorialis L. clones collected from diverse habitats. Hort Res. 3(4):329–332.
- Jing WC, Xu JM, Yang L. 1999. A study on cytotaxonomy of sect. Anguinum of Allium. Acta Phytotaxon Sin. 37:20–34.
- Jones DT, Taylor WR, Thornton JM. 1992. The rapid generation of mutation data matrices from protein sequences. Comput Appl Biosci. 8(3):275–282.
- Kamenetsky R, Faigenboim A, Shemesh-Mayer E, Michael TB, Gershberg C, Kimhi S, Esquira I, Shalom SR, Eshel D, Rabinowitch HD, et al. 2015. Integrated transcriptome catalogue and organ-specific profiling of gene expression in fertile garlic (Allium sativum L.). BMC Genomics. 16:12.
- Kawano S, Nagai Y. 2005. Life-history monographs of Japanese plants. 4: Allium victorialis L. ssp. platyphyllum (Makino) Hulten (Alliaceae) syn. Allium victorialis L. var. platyphyllum Makino; A. latissimum Prokh. Plant Species Biol. 20(3):219–225.
- Khan S, Fatima I, Kazmi MH, Malik A. 2013. New secondary metabolites from Allium victorialis. Helvet Chim Acta. 96(6):1176–1181.
- Khosa JS, Lee R, Bräuning S, Lord J, Pither-Joyce M, McCallum J, Macknight RC. 2016. Doubled haploid 'CUDH2107' as a reference for bulb onion (Allium cepa L.) research: development of a transcriptome catalogue and identification of transcripts associated with male fertility. PLoS One. 11(11):e0166568.
- Kumar S, Stecher G, Li M, Knyaz C, Tamura K. 2018. MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol. 35(6):1547–1549.
- Lee KT, Choi JH, Kim DH, Son KH, Kim WB, Kwon SH, Park HJ. 2001. Constituents and the antitumor principle of Allium victorialis var. platyphyllum. Arch Pharm Res. 24(1):44–50.
- Li P, Su T, Wang H, Zhao X, Wang W, Yu Y, Zhang D, Wen C, Yu S, Zhang F. 2019. Development of a core set of KASP markers for assaying genetic diversity in Brassica rapa subsp. chinensis Makino. Plant Breed. 138(3):309–316.
- Li QQ, Zhou SD, He XJ, Yu Y, Zhang YC, Wei XQ. 2010. Phylogeny and biogeography of Allium (Amaryllidaceae: Allieae) based on nuclear ribosomal internal transcribed spacer and chloroplast rps16 sequences, focusing on the inclusion of species endemic to China. Ann Bot. 106(5):709–733.
- Lim HT, Song YN, You KU, Kim WB. 1998. Evaluation of intraspecific variations of Allium victorialis var. platyphyllum using amplified polymorphic DNA markers. Acta Hortic. 467(467):169–176.
- Liu T, Zeng L, Zhu S, Chen X, Tang Q, Mei S, Tang S. 2015. Large-scale development of expressed sequence tag-derived simple sequence repeat markers by deep transcriptome sequencing in garlic (Allium sativum L.). Mol Breed. 35(11):204.
- Lu Y, Deng YQ, Lu LD, He XJ. 2017. Karyotypes of nineteen populations of four species in Allium subgenus Anguinum. Guihaia. 37:811–821.
- Ma KB, Yang SJ, Jo YS, Kang SS, Nam M. 2021. Development of kompetitive allele specific PCR markers for identification of persimmon varieties using genotyping-by-sequencing. Electron J Biotechnol. 49:72–81.
- Rajkumar H, Ramagoni RK, Anchoju VC, Vankudavath RN, Syed AUZ. 2015. De novo transcriptome analysis of Allium cepa L. (onion) bulb to identify allergens and epitopes. PLoS One. 10(8):e0135387.
- Ryu J, Kim WJ, Im J, Kim SH, Lee KS, Jo HJ, Kim EY, Kang SY, Lee JH, Ha BK. 2018. Genotyping-by-sequencing based single nucleotide polymorphisms enabled kompetitive allele specific PCR marker development in mutant Rubus genotypes. Electron J Biotechnol. 35:57–62.
- Schwarz G. 1978. Estimating the dimension of a model. Ann Stat. 6:461–464.
- Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 30(9):1312–1313.
- Sun X, Zhou S, Meng F, Liu S. 2012. De novo assembly and characterization of the garlic (Allium sativum) bud transcriptome by Illumina sequencing. Plant Cell Rep. 31(10):1823–1828.
- Tang J, Wang FZ. 1980. Flora of China. Beijing: Science Press.
- Tolgor. 1991. A variety of new record and a form of new combination of genus Allium L. from Northeast China. J Jilin Agric Univ. 13:102–103.
- Tolgor, Kang XG, Zhang DR. 1993. Study of Allium in Northeast China. J Jilin Agric Univ. 15:45–48, 107.
- Waterhouse RM, Seppey M, Simão FA, Manni M, Ioannidis P, Klioutchnikov G, Kriventseva EV, Zdobnov EM. 2018. BUSCO applications from quality assessments to gene prediction and phylogenomics. Mol Biol Evol. 35(3):543–548.
- Woo KW, Lee KR. 2013. Phytochemical constituents of Allium victorialis var. platyphyllum. Nat Prod Sci. 19:221–226.
- Yang JK, Kim JS, Jung JY, Jeong MJ, Song HJ, Yun CW, Kim HH, Do ES, Chang JP, Karigar SC, et al. 2014. Habitat influences composition of volatile constituents in Allium victorialis var. platyphyllum. Pak J Bot. 46:271–278.
- Yoo KO, Kim WB, Park HJ, Lim SC, Jang HT. 1998. External morphology and numerical taxonomy among habitat of Allium victorialis var. platyphyllum. Korean J Plant Res. 11:210–216.
- Zhang C, Li X, Zhan Z, Cao L, Zeng A, Chang G, Liang Y. 2018. Transcriptome sequencing and metabolism analysis reveals the role of cyanidin metabolism in dark-red onion (Allium cepa L.) bulbs. Sci Rep. 8(1):14109.
- Zheng X, Tang S, Zhu S, Dai Q, Liu T. 2016. Identification of an NAC transcription factor family by deep transcriptome sequencing in onion (Allium cepa L.). PLoS One. 11(6):e0157871.
- Zhou SM, Chen LM, Liu SQ, Wang XF, Sun XD. 2015. De novo assembly and annotation of the Chinese chive (Allium tuberosum Rottler ex Spr.) transcriptome using the Illumina platform. PLoS One. 10(7):e0133312.
- Zhu S, Chen X, Liu X, Zhao J, Liu T. 2019. Transcriptome-wide association study and eQTL analysis to assess the genetic basis of bulb-yield traits in garlic (Allium sativum). BMC Genomics. 20(1):657.
- Zhu S, Tang S, Tan Z, Yu Y, Dai Q, Liu T. 2017. Comparative transcriptomics provide insight into the morphogenesis and evolution of fistular leaves in Allium. BMC Genomics. 18(1):60.