
Abstract
Arundo formosana Hack. belongs to the Arundionideae subfamily of Poaceae. In this study, we sequenced and assembled the complete chloroplast genome of A. formosana. The complete chloroplast genome was 136,919 bp in size, including a large single copy region of 82,039 bp, a small single-copy region of 12,108 bp and a pair of reverse repeats of 21,386 bp in size. The annotation of A. formosana indicates that it contained 81 protein-coding genes, 47 tRNA and 8 rRNA. Our phylogenetic analysis of the 36 grass complete chroloplast genomes of protein-coding genes using Cyperus rotundus as outgroup showed that A. formosana is closely related to Crinipes species to form the Arundionideae lineage of the grass family.
Arundo formosana Hack. belongs to the Arundionideae subfamily of Poaceae. It is a kind of grass growing on the edge of the coastal rock wall or the grassland on the hillside, 350–450 meters above sea level. The species is endemic in Taiwan and Yunnan provinces of China. Based on five intergenic regions of chloroplast DNA fragments, a phylogeographic study of A. formosana demonstrates the pertinence of infraspecific taxa in integrative taxonomy and phylogeography below the species level (Hardion et al. Citation2017). Indeed, subspecies, varieties, and morphologies are the most common taxonomic levels under the species level (McNeil et al. Citation2012). Recent decades have witnessed extensive efforts to reconstruct the phylogeny of Gramineae. However, the phylogenetic relationships within several clades have not been fully resolved (Clark et al. Citation1995). The analysis of 81 genes from 64 plastid genomes have fully resolved relationships in angiosperms and identified genome-scale evolutionary patterns (Jansen et al. Citation2007). The chloroplast DNAs (cpDNAs) sequences have been increasingly used for resolving the deep phylogeny of plants because of their low rates of nucleotide substitutions and genomic structural changes (Jansen et al. Citation2007). Thus, there is a still great demand to generate the complete chloroplast grass genomes to question about origins and evolution of Poaceae plants on Earth.
In this study, A. formosana plants were collected in the suburbs of Menghai (22°28′32″N, 99°56′30″E), Xishuangbanna State, Yunnan Province, China. The voucher specimen (SCAU 2020148) was deposited in SCAU (the herbarium of the College of Agriculture, South China Agricultural University https://nxy.scau.edu.cn, Li-zhi Gao, [email protected]), China. About 20 g fresh mature leaves were sampled from A. formosana, and cpDNAs were extracted by following a modified high salt method reported formerly (Shi et al. Citation2012). After the cpDNA isolation, approximately 5–10 µg of DNA was sheared, followed by adapter ligation and library amplification, and then subjected to Illumina Sample Preparation Instructions. The fragmented cpDNAs were sequenced at both single-read using the Illumina Genome Analyzer IIx platform at the in-house facility at The Germplasm Bank of Wild Species in Southwestern China, Kunming, China. The obtained paired-end reads (2 × 100 bp read lengths) were assembled using SOAP de novo (Li et al. Citation2010). Regions with ambiguous alignment (conflicted reads mapped to the same genomic region) were trimmed off manually and considered as gaps. Polymerase chain reaction (PCR) amplified fragments yielded by primers derived from the terminal ends of contigs, and the fragments were then sequenced to flank the gap regions. The PCR amplification reactions were template denaturation at 80 °C for 5 min followed by 30 cycles of denaturation at 95 °C for 30 sec, primer annealing at 55 °C for 30 sec, and primer extension at 65 °C for 1 min; followed by a final extension step of 5 min at 65 °C. PCR products were separated by electrophoresis in 1.5% agarose gel and sequenced on an Applied Biosystems (ABI) 3730 automated sequencer. Subsequently, gene prediction and annotation were performed by DOGMA (Wyman et al. Citation2004).
The complete chloroplast genome of A. formosana was 136,919 bp in size, comprising two inverted repeat regions (IRs) with a total of 42,772 bp in size, which were split by a large single copy (LSC) with 82,039 bp and small single copy (SSC) with 12,108 bp in length. The chloroplast genome contained 136 functional genes, including 81 protein-coding genes, 47 tRNAs, and 8 rRNAs. A total of 16 genes were repeated in the IR regions, including 4 rRNA genes (rrn16, rrn23, rrn4.5, and rrn5), 6 protein-coding genes (rpl23, ycf15, ndhB, rps15, rps12 and rps7) and 6 tRNA genes (trnI-CAU, trnL-CAA, trnV-GAU, trnI-GAU, trnR-ACG, and trnN-GUU). The overall GC content of the A. formosana chloroplast genome was ∼38.73% with the corresponding values of 36.87%, 33.12% and 44.93% in the LSC, SSC, and IR regions, respectively.
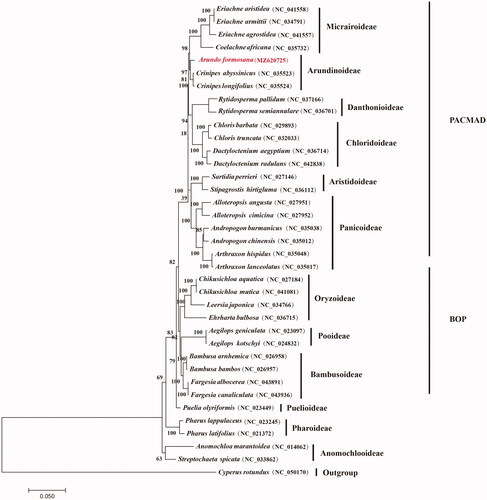
To determine the phylogenetic position of A. formosana in the grass family, 35 grass chloroplast genomes of all protein-coding genes together with Cyperus rotundus from Cyperaceae were separately downloaded from GenBank. Phylogenomic analysis was performed by incorporating the A. formosana chloroplast genome obtained in this study. All sequences were aligned with MAFFT 7.409 (Katoh et al. Citation2002). Using C. rotundus as outgroup phylogenetic tree was reconstructed using the maximum likelihood method using RAxML (Stamatakis Citation2014) based on 1,000 bootstrap replicates. Our results indicated that the 35 examined grass species were evidently clustered into the twelve subfamilies of Poaceae with strong bootstrap supports (). It is apparent that A. formosana is closely related to Crinipes species from Arundionideae of the grass family with strong bootstrap supports.
Figure 1. Maximum likelihood phylogenetic tree based on all protein-coding genes of the 36 grass complete chloroplast genomes using Cyperus rotundus as outgroup. Bootstraps values (1000 replicates) are shown at the nodes.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
The genome sequence data that support the findings of this study are openly available in GenBank of NCBI at [https://www.ncbi.nlm.nih.gov] (https://www.ncbi.nlm.nih.gov/) under the accession no. MZ620725. The associated BioProject, SRA, and Bio-Sample numbers are PRJNA744348, SRR15130727, and SAMN20166948 respectively. The data that newly obtained at this study are also publicly available in the National Genomics Data Center at https://ngdc.cncb.ac.cn under the accession number of GWHBCHV00000000.
Additional information
Funding
References
- Clark LGIS, Zhang W, Wendel JF. 1995. A phylogeny of the grass family (Poaceae) based on ndhF sequence data. Syst Bot. 20(4):436–460.
- Hardion L, Verlaque R, Vorontsova MS, Combroux I, Chen C, Takamizo T, Vila B. 2017. Does infraspecific taxonomy match species evolutionary history? A phylogeographic study of Arundo formosana (Poaceae). Bot J Linn Soc. 183(2):236–249.
- Jansen RK, Cai Z, Raubeson LA, Daniell H, Depamphilis CW, Leebens-Mack J, Muller KF, Guisinger-Bellian M, Haberle RC, Hansen AK, et al. 2007. Analysis of 81 genes from 64 plastid genomes resolves relationships in angiosperms and identifies genome-scale evolutionary patterns. Proc Natl Acad Sci USA. 104(49):19369–19374.
- Katoh K, Misawa K, Kuma K, Miyata T. 2002. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 30(14):3059–3066.
- Li R, Zhu H, Ruan J, Qian W, Fang X, Shi Z, Li Y, Li S, Shan G, Kristiansen K, et al. 2010. De novo assembly of human genomes with massively parallel short read sequencing. Genome Res. 20(2):265–272.
- McNeil J, Barrie FR, Buck WR, Demoulin V, Greuter W, Hawksworth DL, Herendeen PS, Knapp S, Marhold K, Prado J, et al. 2012. International code of nomenclature for algae, fungi, and plants (Melbourne Code). Regnum Vegetabile 154. Königstein: Koelz Scientific Books
- Shi C, Hu N, Huang H, Gao J, Zhao Y, Gao L, Xu Y. 2012. An improved chloroplast DNA extraction procedure for whole plastid genome sequencing. PLOS One. 7(2):e31468.
- Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 30(9):1312–1313.
- Wyman SK, Jansen RK, Boore JL. 2004. Automatic annotation of organellar genomes with DOGMA. Bioinformatics. 20(17):3252–3255.