
Abstract
Gynura cusimbua (D. Don) S. Moore is a favorite food vegetable and traditional folk medicine. The chloroplast genome information, of G. cusimbua, was introduced and released in this study. The complete chloroplast genome was characterized as 156, 684 base pairs (bp) in length. The circle gDNA contained four segments, namely LSC (large single copy), SSC (small single copy) and two IRs (inverted repeats), which was 86, 834 bp and 18, 414 bp and 25, 718 bp in length separately. The total GC content was 36.88%. A total of 125 genes were characterized in the chloroplast genome, where 84, 33 and 8 genes were for coding-proteins, tRNA and rRNA respectively. The phylogeny tree demonstrated that G. cusimbua was clustered with Jacobaea valgaris and Senecio valgaris. This study would fill a vacancy of chloroplast genome information involving G. cusimbua, and provide new genetic resources for the study on Senecioninae.
Introduction
Gynura cusimbua (D. Don) S. Moore 1912 is a favorite food vegetable worldwide. Simultaneously, it was employed as a traditional folk medicine in India and China (Ma et al. Citation2020). The phytochemical studies on G. cusimbua showed the presence of phenylpropanoid glycosides, fat-soluble components and volatiles (Ma et al. Citation2020). However, there were still known rarely on its genetic information. Here, we released the complete sequences of chloroplast genome, which would offer valuable gene information for further studies.
The seeds of G. cusimbua was gifted from Yunnan University, China. Seeds were grew in garden soil and kept in cabinet, as conditions of 26 ± 2 °C, Light/Dark: 16 h/8h, 60% relative humidity, at the Agriculture Station, Zhejiang University (120°5′29″E, 30°17′47″N). Fresh leaves were sampled and subsequently extracted the total DNA via general CTAB method. The specimen, with the accessory number GC-LM002, was deposited in the Museum of CAB, Zhejiang University ([email protected]). The whole-genome sequencing was performed by Novogene biotechnologies Inc. (Tianjin, China) with Illumina Hiseq 2000 platform. A genomic shotgun library with an insertion size of 150 bp was constructed, then the clean data were de novo assembled using NOVOPlasty (Version 3.8.1) (Dierckxsens et al. Citation2017). Finally, the assembled chloroplast genome was annotated by PGA (Qu et al. Citation2019) and GeSeq (Tillich et al. Citation2017).
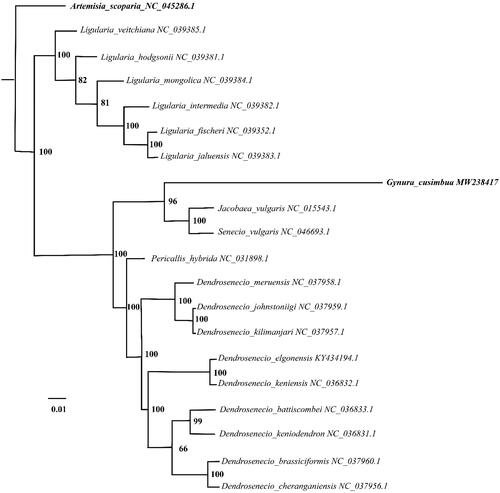
The complete chloroplast genome, of G. cusimbua, was characterized as 156, 684 base pairs (bp) in length. The LSC (large single copy) and SSC (small single copy) is 86, 834 bp and 18, 414 bp respectively, which accounts for 55.42% and 11.75% in the whole circle DNA. Additionally, IRa and IRb shared the same length with 25, 718 bp. The percentage of GC content was 36.88. 125 genes were characterized in the chloroplast genome, in which 84, 33 and 8 genes were for coding-proteins, tRNA and rRNA respectively. To analyze the G. cusimbua phylogenetic position within Senecioninae lineage, 20 chloroplast genomes were employed to align and construct phylogenetic tree by MAFFT (V7.407) (Katoh and Standley Citation2013) and IQtree (Vesion 1.7) (Nguyen et al. Citation2015), correspondingly. The results indicated that G. cusimbua was clustered with Jacobaea valgaris and Senecio valgaris ().
Figure 1. The Maximum-Likelihood (ML) phylogenetic tree of 20 complete chloroplast genomes: Gynura cusimbua is showed with bold italic text for highlight, Artemisia scoparia as an out-group and showed with bold italic text. The numbers adjacent to the nodes denote bootstrap support values from 1000 replicates.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
The genome sequence data that support the findings of this study are openly available in GenBank of NCBI at [https://www.ncbi.nlm.nih.gov] (https://www.ncbi.nlm.nih.gov/) under the accession no. MW238417. The associated BioProject, SRA, and Bio-Sample numbers are PRJNA722038, SRS8704631, and SAMN18743908, respectively.
Additional information
Funding
References
- Dierckxsens N, Mardulyn P, Smits G. 2017. NOVOPlasty: de novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 45(4):e18.
- Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 30(4):772–780.
- Ma Q, Wei R, Zhong G, Sang Z. 2020. Neuroprotective flavonoids from the aerial parts of Gynura cusimbua. Chem Nat Compd. 56(4):725–728.
- Nguyen LT, Schmidt HA, von Haeseler A, Minh BQ. 2015. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. 32(1):268–274.
- Qu XJ, Moore MJ, Li DZ, Yi TS. 2019. PGA: a software package for rapid, accurate, and flexible batch annotation of plastomes. Plant Methods. 15:50.
- Tillich M, Lehwark P, Pellizzer T, Ulbricht-Jones ES, Fischer A, Bock R, Greiner S. 2017. GeSeq – versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 45(W1):W6–W11.