
Abstract
The first complete mitogenome of Trogidae, Omorgus chinensis (Coleoptera: Trogidae) is sequenced using the next generation sequencing. The genomic structure is a circular molecule with 18682 bp in length, comprising 13 protein-coding genes, 22 transfer RNA genes (tRNAs), 2 ribosomal RNAs (rRNAs) and a control region. The nucleotide composition is A (39.44%), C (13.82%), T (36.78%), and G (9.96%) with an AT content of 76.22%. The phylogenetic analysis of 18 insects in Scarabaeoidae show that Omorgus chinensis shares a close ancestry with Lucanidae and Geotrupidae.
Introduction
Omorgus chinensis (Coleoptera: Trogidae) is an important necrophagous insect with an oval shape and tan color. Like most members in Trogidae, this species has a marked engraved and dimpled area on the anterior thoracic plate and a strong arch on the back but a flattened ventral surface (Ren Citation2003). In recent years, with the development of molecular biology, especially the advancement of next-generation sequencing technology, mitochondrial genome (mitogenome) have been widely used as a typical marker for the phylogenetic study of coleopteran insects (Cameron Citation2014; Li et al. Citation2015; Timmermans et al. Citation2015). Fagua et al. Citation2018; However, there are only a few molecule phylogenetic researches in Trogidae (Strümpher et al. Citation2014), and no complete mitogenome data has yet been sequenced.
In this study, we sequenced the complete mitogenome of Omorgus chinensis for the first time using the next-generation methods and reconstructed the phylogenetic relationships within other species in Scarabaeoidae, which provides reliable data for subsequent further phylogenetic studies.
Materials and methods
Sample collection and DNA extraction
The specimens of O. chinensis were collected by Yongjing Chen and Shiju Zhou ([email protected]) from Yaoluoping National Nature Reserve, Anhui province, P. R. China, in July 2017. For adult specimens, the muscle tissues were placed in 100% ethanol. Total genomic DNA was extracted from the muscle of a single O. chinensis using the Qiagen DNAeasy Kit. The specimen deposited in the Museum of Anhui university and the sequence was submitted to GenBank with the accession number MK937809.
Polymerase chain reaction amplification, sequencing and assembly
Complete mitogenome was assembled by overlapping high-throughput fragment sequences, the amplification of the three primers were used as markers for tandem high-throughput sequencing fragment sequences. The primers used for amplification listed in . PCR amplification reactions were carried out in 25 μL volumes containing 10 μM of each primer (forward and reverse) 1 μL, 2 μL template DNA, 12.5 μL 2 × EasyTaq SuperMix (+dye), and 8.5 μL sterile double-distilled water to make up a final volume of 25 μL. The polymerase chain reaction amplifications were performed under the following conditions: an initial denaturation at 94 °C for 2 min, followed by 35–37 cycles of denaturation at 94 °C for 40 seconds, annealing at 52–58 °C for 50 seconds, and elongation at 70 °C for 1 min, and then a final extension step at 72 °C for 7 min. The temperature of annealing was determined by the length of fragments. An Illumina TruSeq library was prepared with an average insert size of 450 bp and was sequenced using the Illumina Hiseq2500 platform with 250 bp paired-end reads. Raw reads were trimmed of adapters using Trimmomatic. Low quality and short reads were removed with Prinseq. High quality reads were used in denovo assembly using IDBA-UD with minimum and maximum k values of 80 and 240 bp, respectively. To investigate the accuracy of the assembly, clean reads were mapped onto the obtained mt contig using Geneious Prime 2019.1.1 (http://www.geneious.com/), allowing for mismatches of up to 2%, maximum gap size of 3 bp and minimum overlap of 100 bp. Finally, we obtained the mitogenome of O. chinensis with the average sequencing depth of 242×.
Table 1. Details on primers used in this study.
Mitogenome annotation and analysis
Preliminary annotations made with the MITOS WebServer (Bernt et al. Citation2013) (http://mitos.bioinf.uni-leipzig.de/index.py). tRNA genes and their secondary structures were inferred using tRNAscan-SE 1.21 (Schattner et al. Citation2005) (http://lowelab.ucsc.edu/tRNAscan-SE/). Those not identified by tRNAscan-SE, in addition to 16S ribosomal RNA (rrnL, lrRNA), and 12S ribosomal RNA (rrnS, srRNA), were determined according to sequence similarity with related species. The protein-coding genes (PCGs) were determined by ORF Finder (http://www.ncbi.nlm.nih.gov/gorf/gorf.html) under the invertebrate mitochondrial genetic code. Nucleotide compositions, codon usage, and relative synonymous codon usage (RSCU) values of PCGs were calculated with MEGA-X (Kumar et al. Citation2018). PCGs were translated with DNAMAN v7.0.2.176 (Lynnon Biosoft, Vaudreuil-Dorion, Canada). Composition skew analysis was conducted according to formulas AT-skew = [A − T]/[A + T] and GC-skew = [G − C]/[G + C] (Perna and Kocher Citation1995).
Phylogenetic analyses
A total of 18 mitochondrial genomes of Scarabaeoidae were used for phylogenetic analysis (). Each PCGs was aligned individually based on codon-based multiple alignments using the MEGA-X (Kumar et al. Citation2018). Models of nucleotide substitution were selected according to the Akaike Information Criterion (AIC) with jModelTest v2.1.4 (Posada Citation2008). Phylogenetic trees were generated from ML analysis using RAxML (Stamatakis Citation2014) and Bayesian inference (BI) with MrBayes v3.2.5 (Ronquist et al. Citation2012), both under the GTR + I + G model. Node support in the ML tree was estimated through bootstrap analysis with 1,000 replicates. The BI was conducted with two simultaneous Markov chain Monte Carlo runs of 2 × 109 generations, sampled every 1,000 steps, with the first 25% discarded as burn-in. Phylogenetic trees were viewed and edited in Figtree v1.4.3 (Kim et al. Citation2021) (http://tree.bio.ed.ac.uk/software/figtree/).
Table 2. Taxa used in this study.
Table 3. Mitogenome organization of Omorgus chinensis.
Results and discussion
The mitogenome of O. chinensis is 18,682 bp in length, with the typical gene content as other known Coleoptera mitogenomes: 22 transfer RNA genes (tRNAs), 2 ribosomal RNA genes (16S rRNA and 12S rRNA), 13 protein-coding genes (PCGs) and 1 non-coding AT-rich region (Sheffield et al. Citation2009; Kim and Farrell Citation2015; Wu et al. Citation2016; Yang et al. Citation2016; Du et al. Citation2017). The base compositions of the mitogenome nucleotide composition were A (39.44%), C (13.82%), T (36.78%), and G (9.96%) with an AT content of 76.22%. AT-skew and GC-skews were calculated as 0.035 and −0.162. AT-biased were found in the protein-coding region (75.66%), rRNA (80.09%), tRNAs (78.11%) and control region (75.16%). As a typical invertebrate mitogenome, 12 PCGs of the sequence employ same initiation codons ATN, except the start codon of COI is AAT. 10/13 PCGs shared the typical termination codons TAA and TAG, while others use TA residue or a single T as the terminator codons. All of the 22 tRNAs range from 63 to 70 bp in length, 14 of the 22 tRNA-coding genes were located on the J-strand and others were located on the N-strand (). Secondary structures predicted by the tRNA scan-SE suggested that all the tRNA genes in O. chinensis adopted a typical clover-leaf structurewith the exception of tRNA-Ser (AGN), tRNA-Ser (AGN) has the deficiency of the dihydrouridine arm. which is a typical feature of metazoan mitochondrial genomes (Cameron Citation2014). The control region is 4006 bp long and is located between 12S rRNA and tRNA-Ile. ML and BI analyses obtained the consistent topology. The resolution of maximum likelihood and Bayesian analyses () both indicates that the newly sequenced species O. chinensis (Coleoptera: Trogidae) shared a close ancestor with Anoplotrupes stercorosus (Coleoptera: Geotrupidae) and closely related to Lucanidae.
Figure 1. The ML and BI phylogenetic trees of Omorgus chinensis and 17 other scarabs based on 13 PCGs.
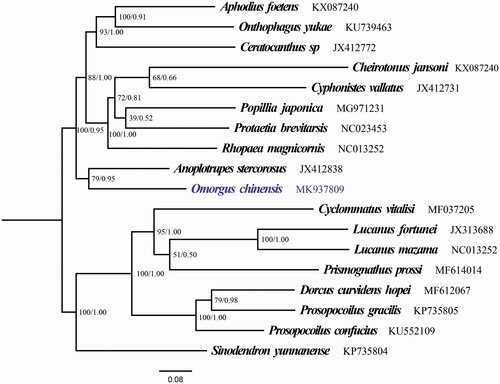
In conclusion, the first complete mitogenome of Trogidae, O. chinensis is a typical metazoan mitogenome. Phylogenetic analysis show that Trogidae shares a close ancestry with Lucanidae and Geotrupidae.
Acknowledgements
We would like to thank the colleague of Professor Weihai Li for his kindly help about the data analysis. We also thank Shiju Zhou for their assistance in this study.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
The genome sequence data that support the findings of this study are openly available in GenBank of NCBI at [https://www.ncbi.nlm.nih.gov] (https://www.ncbi.nlm.nih.gov/) under the accession MK937809. The associated BioProject, Bio-Sample numbers and SAR number are PRJNA759651, SAMN21181119 and SRR 16510902, respectively.
Additional information
Funding
References
- Bernt M, Donath A, Jühling F, Externbrink F, Florentz C, Fritzsch G, Pütz J, Middendorf M, Stadler PF. 2013. MITOS: improved de novo metazoan mitochondrial genome annotation. Mol Phylogenet Evol. 69(2):313–319.
- Cameron SL. 2014. Insect mitochondrial genomics: implications for evolution and phylogeny. Annu Rev Entomol. 59(1):95–117.
- Du C, Zhang L, Lu T, Ma J, Zeng C, Yue B, Zhang X. 2017. Mitochondrial genomes of blister beetles (Coleoptera, Meloidae) and two large intergenic spacers in Hycleus genera. BMC Genomics. 18(1):698.
- Fagua G, Condamine FL, Brunet BMT, Clamens A-L, Laroche J, Levesque RC, Cusson M, Sperling FAH. 2018. Convergent herbivory on conifers by Choristoneura moths after boreal forest formation. Mol Phylogenet Evol. 123:35–43.
- Kim JI, Do TD, Choi Y, Yeo Y, Kim CB. 2021. Characterization and comparative analysis of complete mitogenomes of three Cacatua Parrots (Psittaciformes: Cacatuidae). Genes. 12(2):209.
- Kim SI, Farrell BD. 2015. Phylogeny of world stag beetles (Coleoptera: Lucanidae) reveals a Gondwanan origin of Darwin's stag beetle. Mol Phylogenet Evol. 86:35–48.
- Kumar S, Stecher G, Li M, Knyaz C, Tamura K. 2018. MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol. 35(6):1547–1549.
- Li H, Shao RF, Song N, Song F, Jiang P, Li ZH, Cai WZ. 2015. Higher-level phylogeny of paraneopteran insects inferred from mitochondrial genome sequences. Sci Rep. 5(1):1–10.
- Perna NT, Kocher TD. 1995. Patterns of nucleotide composition at fourfold degenerate sites of animal mitochondrial genomes. J Mol Evol. 41(3):353–358.
- Posada D. 2008. jModelTest: phylogenetic model averaging. Mol Biol Evol. 25(7):1253–1256.
- Ren GD. 2003. Taxonomic studies of the family Trogidae (Coleoptera: Scarabaeoidea) from China. Entomotaxonomia. 25(2):109–117.
- Ronquist F, Teslenko M, van der Mark P, Ayres DL, Darling A, Höhna S, Larget B, Liu L, Suchard MA, Huelsenbeck JP. 2012. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol. 61(3):539–542.
- Schattner P, Brooks AN, Lowe TM. 2005. The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Res. 33(Web Server):W686–9.
- Sheffield NC, Song HJ, Cameron SL, Whiting MF. 2009. Non stationary evolution and compositional heterogeneity in beetle mitochondrial phylogenomics. Syst Biol. 58(4):381–394.
- Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 30(9):1312–1313.
- Strümpher WP, Sole CL, Villet MH, Scholtz C. 2014. Phylogeny of the family Trogidae (Coleoptera: Scarabaeoidea) inferred from mitochondrial and nuclear ribosomal DNA sequence data. Syst Entomol. 39(3):548–562.
- Timmermans MJTN, Barton C, Haran J, Ahrens D, Culverwell CL, Ollikainen A, Dodsworth S, Foster PG, Bocak L, Vogler AP. 2015. Family-level sampling of mitochondrial genomes in Coleoptera: compositional heterogeneity and phylogenetics. Genome Biol Evol. 8(1):161–175.
- Wu YY, Cao YY, Fang J, Wan X. 2016. The first complete mitochondrial genome of stag beetle from China, Prosopocoilus gracilis (Coleoptera, Lucanidae). Mitochondrial DNA A DNA Mapp Seq Anal. 27(4):2633–2634.
- Yang J, Ye F, Huang Y. 2016. Mitochondrial genomes of four katydids (Orthoptera: Phaneropteridae): new gene rearrangements and their phylogenetic implications. Gene. 575(2 Pt 3):702–711.