
Abstract
In this study, we reported the complete chloroplast genome sequence of Clivia robusta for the first time. The complete chloroplast genome of C. robusta was 157,130 bp in length, containing a large single-copy region (LSC, 85,430 bp), a small single-copy region (SSC, 18,278 bp), and two inverted repeat regions (IRs, 26,711 bp). The overall GC content was 38.01%. The chloroplast genome contained 128 genes in total, including 86 protein-coding, 34 tRNA, and eight rRNA genes. The phylogenetic tree showed that C. robusta formed a monophyletic clade with other Clivia species.
Clivia robusta B.G. Murray 2004 belongs to genus Clivia in the family Amaryllidaceae. It is an evergreen herbaceous plant, flowering from late March to early August. Its flowers are pendulous and tubular, orange with green tips (Murray et al. Citation2004). Due to its high ornamental value, we sequenced and analyzed the complete chloroplast genome of C. robusta for the first time, in order to better understand its relationship with related species and provide a scientific basis for hybridization and breeding.
Young fresh leaves of C. robusta were sampled from Liaoning Academy of Agricultural Sciences (N41°48’33”, E123°34’53”), Shenyang, China. Total genomic DNA was isolated following the modified CTAB method for constructing a 400 bp shotgun library, and a specimen was deposited at the Institute of Floriculture of Liaoning Academy of Agricultural Sciences (www.laas.cn, contact person XH Zhao, and email [email protected]) under the voucher number ZZJZL03. The complete chloroplast genome of C. robusta was sequenced on the Illumina NovaSeq platform, assembled with GetOrganelle v1.6.2e (Jin et al. Citation2020), and annotated with the OGAP pipeline (https://github.com/zhangrengang/OGAP). The draft annotations were then adjusted manually. The annotated complete chloroplast genome of C. robusta was deposited in GenBank with accession number MW660367. Phylogenetic tree was generated by maximum likelihood (ML) analysis. The complete chloroplast genomes of 23 representative species of the family Amaryllidaceae were selected, using Asparagus officinalis (Asparagaceae) as an outgroup. The sequences were aligned using MAFFT v7.471 (Standley and Katoh Citation2013), and the multiple alignment was trimmed with trimAl v1.2 (Capella-Gutierrez et al. Citation2009). The phylogenetic tree was reconstructed with the software IQ-TREE v1.6.5 (Nguyen et al. Citation2015). The best-fit K3Pu + F+R3 model was chosen according to the Bayesian information criterion (BIC) using ModelFinder (Kalyaanamoorthy et al. Citation2017). Ultrafast bootstrap (Hoang et al. Citation2018) was used to test branch support values with 1000 replicates.
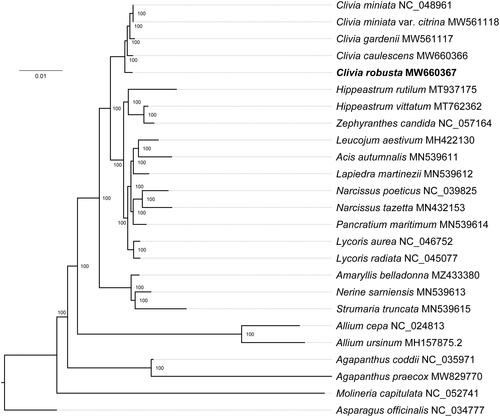
The complete chloroplast genome of C. robusta was 157,130 bp in length and displayed a typical quadripartite structure, which contained a pair of inverted repeat regions (IRs, 26,711 bp) separated by a large single-copy region (LSC, 85,430 bp) and a small single-copy region (SSC, 18,278 bp). The overall GC content was 38.01%. The chloroplast genome of C. robusta contained 128 genes, including 86 protein-coding, 34 tRNA, and eight rRNA genes. There were 19 genes duplicated in the IR regions, including 7 protein-coding genes, eight tRNA, and four rRNA genes. The phylogenetic tree showed that C. robusta and other Clivia species formed a monophyletic clade within the family Amaryllidaceae with 100% bootstrap values (). The degrees of differentiation between C. robusta and other Clivia species (C. caulescens, C. gardenii, C. miniata, and C. miniata var. citrina) were 0.41%, 0.44%, 0.40%, and 0.40%, respectively. These results can provide genomic resources for the breeding as well as speciation of Clivia species.
Figure 1. Maximum likelihood (ML) tree based on the complete chloroplast genome sequences of C. robusta and other 23 species, using Asparagus officinalis (Asparagaceae) as the outgroup. Bootstrap values are indicated at the nodes.
Disclosure statement
No potential conflict of interest was declared by the author(s).
Data availability statement
The data that support the analysis and results of this study are openly available in Genbank with accession number (MW660367) (http://www.ncbi.nlm.nih.gov/). The associated BioProject, SRA, and BioSample numbers are PRJNA704719, SRR13781249, and SAMN18053628, respectively.
Additional information
Funding
References
- Capella-Gutierrez S, Silla-Martinez JM, Gabaldon T. 2009. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics. 25(15):1972–1973.
- Hoang DT, Chernomor O, von Haeseler A, Minh BQ, Vinh LS. 2018. UFBoot2: improving the ultrafast bootstrap approximation. Mol Biol Evol. 35(2):518–522.
- Jin J-J, Yu W-B, Yang J-B, Song Y, dePamphilis CW, Yi T-S, Li D-Z. 2020. GetOrganelle: a fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 21(1):241.
- Kalyaanamoorthy S, Minh BQ, Wong TKF, von Haeseler A, Jermiin LS. 2017. ModelFinder: fast model selection for accurate phylogenetic estimates. Nat Methods. 14(6):587–589.
- Murray BG, Ran Y, DE Lange PJ, Hammett KRW, Truter JT, Swanevelder ZH. 2004. A new species of Clivia (Amaryllidaceae) endemic to the Pondoland Centre of Endemism, South Africa. Bot. J. Linn. Soc. 146(3):369–374.
- Nguyen L-T, Schmidt HA, von Haeseler A, Minh BQ. 2015. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. 32(1):268–274.
- Standley DM, Katoh K. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 30(4):772–780.