
Abstract
In this study, we sequenced and analyzed the complete mitogenome Dahira obliquifascia to compare mitogenomic structures and reconstruct phylogenetic relationships. The complete mitogenome sequence of D. obliquifascia is circular, 15,939 bp in size and encodes 13 protein-coding genes (PCGs), 2 ribosomal RNA genes (rRNA), 22 transfer RNA genes (tRNA) and a control region (CR). Nucleotide composition is highly biased toward A + T nucleotides (80.3%). All 13 protein-coding genes (PCGs) initiate with the standard start codon of ATN and terminate with the typical stop codon TAA/TAG. Phylogenetic analyses were performed using 13 protein coding genes (PCGs) showed that D. obliquifascia is closely related to Theretra oldenlandiae.
Dahira obliquifascia Hampson, 1910 (Lepidoptera: Sphingidae) is a kind of hawkmoth which is widely distributed in Southeast Asia. The mitogenome sequence of D. obliquifascia so far remains unknown. Therefore, we sequenced the complete mitochondrial DNA genome of D. obliquifascia to provide more comprehensive data for this species and reconstructed the phylogenetic relationship of Sphingidae.
Dahira obliquifascia was collected from the Menghai County, Yunnan Province, China (100°03′24″, 21°41′53″), April 17 of 2021, specimens and DNA were deposited in the Entomological Museum, College of Life Sciences, Anhui Normal University (https://www.ahnu.edu.cn/, YX, Huang, [email protected]) under the accession no. YN20210418. All animal-related experiments were performed according to the protocols approved by the Institutional Animal Care and Use Committee of Anhui Normal University (Grant number AHNU-ET2021032). A whole genome shotgun (WGS) strategy was used after the exacted total DNA was quantified. Illumina Novaseq S6000 platform was employed for sequencing with a strategy of 150 paired-ends (PE) (Andrews Citation2020). The raw paired reads were quality-trimmed and assembled into the complete circular mitogenome in Novoplasty 2.7.2.
The mitogenome sequence data of D. obliquifascia is available in the GenBank (https://www.ncbi.nlm.nih.gov/) under the accession number MZ343807. The mitogenome of D. obliquifascia is 15,939 bp in size and includes 13 PCGs, 22 tRNAs, 2 rRNAs and a control region (Cameron Citation2014). The composition of D. obliquifascia is similar to many insect mitogenomes reported previously. D. obliquifascia mitochondrial genomes had a total length of 15,939, which were typical double-chain circular molecular structures. The average nucleotide composition of overall base composition of the mitogenome was A: 41.3%, T: 39%, C: 12.4% and G: 7.3%. The nucleotide composition of all the mitogenomes had a high A + T content, with an average of 80.3%, showing a strong A/T bias. In D. obliquifascia, 24 genes (15 tRNAs and nine PCGs) were encoded by the majority-strand (J-strand) and 13 genes (4 PCGs, 2 rRNAs and 7 tRNAs) were encoded by the minority- strand (N-strand). Among the 13 PCGs in the D. obliquifascia mitogenome, 9 PCGs (nad2, nad3, nad6, cox1, cox2, cox3, atp6, atp8,cytb) are encoded by the J strand, while the other 4 PCGs are encoded on the N strand. All 13 PCGs start with ATN and stop with traditional TAA or TAG codons, which is similar to most other insect mitogenomes (Crozier and Crozier Citation1993; Korkmaz et al. Citation2015).
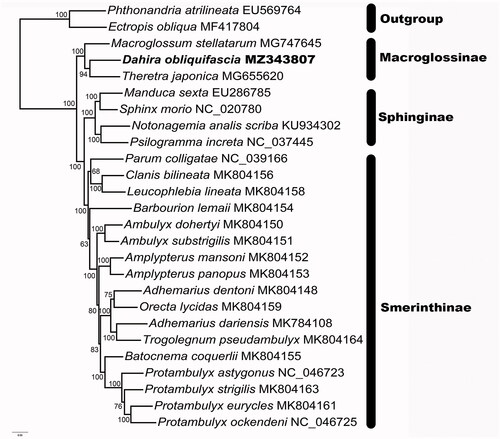
Tweenty six Sphingidae species were selected to reconstructed the phylogeny of Sphingidae. Each protein coding genes were aligned by MAFFT (Katoh et al. Citation2005). W-IQ-Tree web server was used to reconstruct the phylogeny under the ML (maximum-likelihood) method (Trifinopoulos et al. Citation2016). The result was clearly exhibited that all the subfamilies of Sphingidae were monophyly, which was in accordance with previous study (Wang et al. Citation2021). Smerinthinae was the most derived group in Sphingidae, and the most closely related with Sphinginae (). Macroglossinae was recovered as sister to the clade formed by Smerinthinae and Sphinginae. In Macroglossinae, Macroglossum stellatarum was first diverged. D. obliquifascia is closely related to Theretra oldenlandiae.
Figure 1. Phylogenetic relationships within Sphingidae based on 13 protein-coding genes were performed using ML methods.
Credit authorship statement
Li-Qing Qi: methodology, writing – original draft. Hong Zhang: methodology. Xu Wang: conceptualization, funding acquisition. Yi-Xin Huang: conceptualization, funding acquisition. All authors agree to be accountable for all aspects of the work.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
The data that support the findings of this study are openly available in GenBank at https://www.ncbi.nlm.nih.gov/genbank/, reference number MZ343807. The associated BioProject, Bio-Sample numbers, and SRA are PRJNA735332, SAMN19579703, and SRR14741682, respectively.
Additional information
Funding
References
- Andrews S. 2020. FastQC: a quality control tool for high throughput sequence data. http://www.bioinformatics.babraham.ac.uk/projects/fastqc [accessed 2020 Jul 10].
- Cameron SL. 2014. Insect mitochondrial genomics: implications for evolution and phylogeny. Annu Rev Entomol. 59:95–117.
- Crozier RH, Crozier YC. 1993. The mitochondrial genome of the honeybee Apis mellifera: complete sequence and genome organization. Genetics. 133(1):97–117.
- Katoh K, Kuma KI, Toh H, Miyata T. 2005. MAFFT version 5: improvement in accuracy of multiple sequence alignment. Nucleic Acids Res. 33(2):511–518.
- Korkmaz EM, Doğan Ö, Budak M, Başıbüyük HH. 2015. Two nearly complete mitogenomes of wheat stem borers, Cephus pygmeus (L.) and Cephus sareptanus Dovnar-Zapolskij (Hymenoptera: Cephidae): An unusual elongation of rrnS gene. Gene. 558(2):254–264.
- Trifinopoulos J, Nguyen L-T, Von Haeseler A, Minh BQ. 2016. W-IQ-TREE: a fast online phylogenetic tool for maximum likelihood analysis. Nucleic Acids Res. 44(W1):W232–W235. doi:https://doi.org/10.1093/nar/gkw256.
- Wang X, Zhang H, Kitching I, Xu ZB, Huang YX. 2021. First mitogenome of subfamily Langiinae (Lepidoptera: Sphingidae) with its phylogenetic implications. Gene. 789:145667.