
Abstract
Peucedanum hakuunense Nakai is one of the rare species in the Korean Peninsula. This study characterized the complete plastid genome (plastome) sequence of P. hakuunense by de novo assembly with next-generation sequencing data. The complete plastome of P. hakuunense is 147,426 bp in length with a typical quadripartite structure comprising a large single-copy region of 91,915 bp, a small single-copy region of 17,425 bp, and two inverted repeat regions of 19,043 bp in length. The plastome of P. hakuunense is composed of 85 protein-coding genes, 36 tRNA genes, and 8 rRNA genes. The phylogenetic analysis revealed that two Peucedanum species formed an independent subclade, sister to the subclade of Angelica species within the tribe Selineae.
Peucedanum hakuunense Nakai (common name: Baekun hog fennel) is a member of the genus Peucedanum distributed only in the Korean Peninsula. This species was first reported by Nakai (1939) (Lee Citation1982) has a morphological characteristic of 30 ∼ 40 cm in plant height, with a narrow pinnate tri-ternate radical leaf of 10–18 cm in length with a wide triangle lobe bipinnate leaf. It has a compound umbel with 15–30 individual white flower stalks with purple stamen. Mericarp fruit is of flat oval shape with 4.5 mm length and 2.5 mm width with three veins on the back with white wings on the edge. Of all, the Peucedanum species discovered within the Korean Peninsula, P. hakuunense is one of the rare species (Na et al. Citation2017). Although it was considered an endemic and rare species in Korea, P. hakuunense was poorly studied in genetic and genomic approaches. Here we report the complete plastid genome sequence of P. hakuunense to facilitate taxonomic and phylogenetic approaches from phylogenetic inference on the genus Peucedanum and advance the understanding of the phylogenetic relationship in the family Apiaceae.
Peucedanum hakuunense specimen was collected from Baekun Mountain located in Kwangyang City, Jeollanam-do Province (35°1′42″ N, 127°36′13″ E, Nambu University Forest, Seoul National University). DNA was extracted using the CTAB protocol (Allen et al. Citation2006). The specimen's isolated genomic DNA was deposited at the National Institute of Biological Resources (42, Hwangyeong-ro, Seo-gu, Incheon 22689, Korea; Contact person: Yoon-Jeong Park, [email protected]) under the specimen number of NIBRGR0000635909. About 1 μg of purified DNA was sequenced using the Illumina Miseq platform (Illumina Inc, San Diego, CA, United States). Approximately 1.2 Gbps paired-end sequencing data were obtained for P. hakuunense sample. High-quality reads were used for de novo assembly on the plastome with low coverage whole genome sequencing (dnaLCW) method (Kim et al. Citation2015). In brief, the raw reads were trimmed using a trimming tool and then assembled into contigs using the CLC assembly tool (ver. 4.06 beta, CLC Inc, Aarhus, Denmark). Using a previously reported plastome sequence of Peucedanum japonicum (NC_034644) as a reference, the assembled contigs with high similarity to the reference sequence were then extracted using MUMmer (Kurtz et al. Citation2004) and eventually assembled into a whole plastome sequence. Gene annotation on the draft plastome sequence of P. hakuunense was performed using GeSeq (Tillich et al. Citation2017) and Artemis (Rutherford et al. Citation2000) and then manually curated.
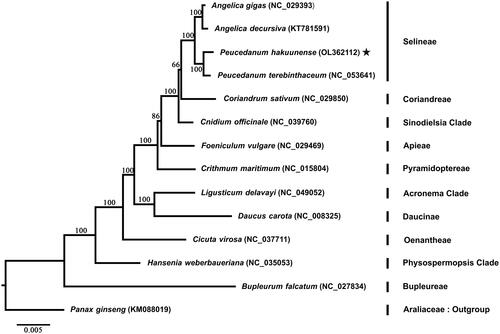
The plastome of P. hakuunense was 147,426 bp in length with 37.52% of GC content. The plastome structure was typical quadripartite featuring two copies of inverted regions (IRa and IRb) with 19,043 bp each, a large single-copy region with 91,915 bp, and a small single-copy region with 17,425 bp in length. A total of 129 genes, including 85 protein-coding genes, 36 tRNA genes, and 8 rRNA genes, were found in the plastome of P. hakuunense. The phylogenetic analysis was performed with representative species in the family Apiaceae. The plastome sequences of 12 Apiaceae species were downloaded from NCBI GenBank (https://www.ncbi.nlm.nih.gov/genbank/), as well as a complete plastome sequence of Panax ginseng in Araliaceae was included as an outgroup. A total of 78 plastid genes, shared by all 14 species, were used for phylogenetic reconstruction (). Each CDS region of plastid genes was extracted by Feature Extract (Wernersson Citation2005) and then concatenated into a single matrix. The maximum-likelihood (Chumley et al. Citation2006) tree was reconstructed using raxmlGUI 2.0 (Edler et al. Citation2021) with 1000 bootstrap replicates under the GTR + G+I model selected by jModeltest v. 2.1.5 (Darriba et al. Citation2012). As a result, the ML tree has demonstrated that P. hakuunenese was sister to Peucedanum terebinthaceum, followed by the subclade of Angelica species (A. gigas and A. decursiva) forming a clade under the tribe Selineae. The phylogenetic relationships within the subfamily Apioideae were consistent with the phylogenetic relationships in a previous study (Wen et al. Citation2021). The newly reported plastid genome sequence of P. hakuunense will be helpful to advance our current understanding of phylogenetic relationships between the genus Peucedanum and the family Apiaceae.
Figure 1. Phylogenetic analysis of Apiaceae based on 78 plastid genes from 14 species. Star indicates the newly generated plastome of P. hakuuense in this study. A phylogenetic tree was reconstructed using the maximum-likelihood method in raxmlGUI 2.0 with 1000 bootstrap replicates. Numbers at the node are the bootstrap supporting value calculated from the ML method. The plastome sequence of Panax ginseng from Araliaceae was included as an outgroup.
Ethical approval
The plant specimen is not designated as endangered species; therefore, it requires no specific permissions or licenses.
Authors’ contributions
Analysis and interpretation of the data: Ho Jun Joh; Drafting of the paper: Ho Jun Joh; Revising: Hyun-Seung Park, Jong-Soo Kang, Jee Young Park, and Tae-Jin Yang.
Acknowledgments
We want to express our gratitude to the National Institute of Biological Resources for allowing us to deposit our DNA sample.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
The plastid genome sequence data supporting this study’s finding is available in NCBI GenBank under the accession number OL362112. The associated BioProject, SRA, and Bio-sample numbers are PRJNA775900, SRR16611559, and SAMN22635847, respectively.
Additional information
Funding
References
- Allen G, Flores-Vergara M, Krasynanski S, Kumar S, Thompson W. 2006. A modified protocol for rapid DNA isolation from plant tissues using cetyltrimethylammonium bromide. Nat Protoc. 1(5):2320–2325.
- Chumley TW, Palmer JD, Mower JP, Fourcade HM, Calie PJ, Boore JL, Jansen RK. 2006. The complete chloroplast genome sequence of Pelargonium × hortorum: organization and evolution of the largest and most highly rearranged chloroplast genome of land plants. Mol Biol Evol. 23(11):2175–2190.
- Darriba D, Taboada GL, Doallo R, Posada D. 2012. jModelTest 2: more models, new heuristics and parallel computing. Nat Methods. 9(8):772.
- Edler D, Klein J, Antonelli A, Silvestro D. 2021. raxmlGUI 2.0: a graphical interface and toolkit for phylogenetic analyses using RAxML. Methods Ecol Evol. 12(2):373–377.
- Kim K, Lee S-C, Lee J, Yu Y, Yang K, Choi B-S, Koh H-J, Waminal NE, Choi H-I, Kim N-H. 2015. Complete chloroplast and ribosomal sequences for 30 accessions elucidate evolution of Oryza AA genome species. Sci Rep. 5:15655.
- Kurtz S, Phillippy A, Delcher AL, Smoot M, Shumway M, Antonescu C, Salzberg SL. 2004. Versatile and open software for comparing large genomes. Genome Biol. 5(2):R12–R19.
- Lee TB. 1982. Endemic plants and their distribution in Korea. J Nat Acad Sci. 21:169.
- Na N-R, Kim Y-Y, Lee G-r, Song H-I, Park J-M, Jang C. 2017. Floristic study of Mt. Deoktaesan, Jinan, Jeonbuk. Kor J Plant Res. 30(4):378–398.
- Rutherford K, Parkhill J, Crook J, Horsnell T, Rice P, Rajandream M-A, Barrell B. 2000. Artemis: sequence visualization and annotation. Bioinformatics. 16(10):944–945.
- Tillich M, Lehwark P, Pellizzer T, Ulbricht-Jones ES, Fischer A, Bock R, Greiner S. 2017. GeSeq – versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 45(W1):W6–W11.
- Wen J, Xie D-F, Price M, Ren T, Deng Y-Q, Gui L-J, Guo X-L, He X-J. 2021. Backbone phylogeny and evolution of Apioideae (Apiaceae): new insights from phylogenomic analyses of plastome data. Mol Phylogenet Evol. 161:107183.
- Wernersson R. 2005. FeatureExtract-extraction of sequence annotation made easy. Nucleic Acids Res. 33:W567–W569.