
Abstract
Picrasma quassioides is a member of the Simaroubaceae family and is widely used as a medicinal plant. In this study, we sequenced and assembled the complete chloroplast genome of P. quassioides. The chloroplast genome is 160,015 bp in length, with a large single-copy region of 87,136 bp, a small single-copy region of 18,069 bp, and a pair of inverted repeat regions of 27,405 bp. It contains a total of 110 unique genes, including 77 protein-coding genes, 29 tRNA genes, and 4 rRNA genes. Phylogenetic analysis showed that P. quassioides clustered well with Simaroubaceae plants, Eurycoma longifolia, Leitneria floridana, and Ailanthus latissimus.
Picrasma quassioides (D. Don) Benn. 1844, also known as ‘nigaki’ bitterwood, is a kind of small tree or shrub that belongs to the Simaroubaceae family, which is mainly distributed in tropical and subtropical Asia (Alves et al. Citation2014). As a traditional Chinese medicinal plant, P. quassioides has anti-inflammatory, anti-malarial, anti-cancer, and antihypertensive properties (Bai et al. Citation2020). The crude extract of P. quassioides contains a variety of beneficial phytochemicals, such as alkaloids and polyphenols, which have a wide range of pharmacological activities, from anti-inflammatory to anti-parasitic. So far, most studies on P. quassioides have focused on its chemical composition and pathology, with little attention paid to its molecular biology and evolution. Therefore, in this study, we sequenced, assembled, annotated, and analyzed the phylogenetic tree of the complete chloroplast genome of P. quassioides to increase our understanding of the phylogenetic and evolutionary history of the species.
The green leaves of P. quassioides were collected from Yulin Normal University (110.183°E, 22.660°N) in Yulin, Guangxi, China. The voucher specimen (KM-YNU-001) was deposited in the herbarium of Yulin Normal University (https://syy.ylu.cn/index.html, Yulin Zhu, [email protected]). All the research met ethical guidelines and adhered to the legal requirements of the study country. An enhanced CTAB technique was applied to extract total genomic DNA. The Illumina Hiseq 4000 platform (150 bp * 2) (Biozeron, Shanghai, China) was utilized for the sequencing. Trimmomatic (Bolger et al. Citation2014) was used to remove adapters and low-quality reads from the raw data using the following settings: ILLUMINACLIP 2:30:10, LEADING:3, TRAILING:3, SLIDINGWINDOW:4:25, and MINLEN:85. The GetOrganelle toolkit (Jin et al. Citation2020) and CPGAVAS2 (Shi et al. Citation2019) were used to accomplish the de novo assembly and chloroplast genome annotation using the default parameters. The cp sequence of P. quassioides was submitted to NCBI, and the accession number was MZ902043.
The chloroplast genome of P. quassioides is 160,015 bp in length, with a large single-copy region (LSC) of 87,136 bp, a small single-copy region (SSC) of 18,069 bp, and two inverted repeat regions (IRs) of 27,405 bp. The overall GC content of the P. quassioides cp genome was 37.96%, with the LSC, SSC, and IR regions accounting for 36.16%, 32.39%, and 42.67%, respectively. The cp genome of P. quassioides had 110 distinct genes, including 77 protein-coding genes, 29 tRNA genes, and 4 rRNA genes. Eighteen of these 110 genes had introns, including eight tRNA genes and ten protein-coding genes. The genes ycf3 and clpp had two introns, while the remaining 16 genes only had one intron.
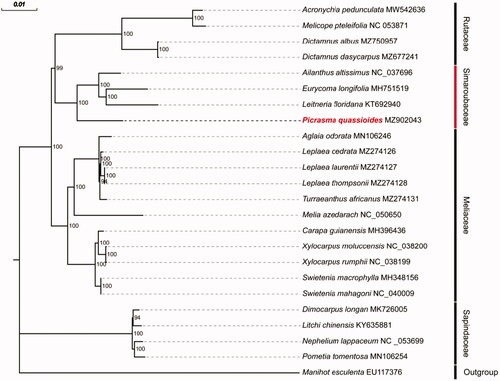
To confirm the phylogenetic position of P. quassioides and further clarify the evolutionary relationships within the family Simaroubaceae, phylogenetic analyses based on a total of 23 complete cp genomes that belong to the order Sapindales were performed. Manihot esculenta, which belongs to the family Euphorbiaceae, was selected as the outgroup. After aligning the cp genomes with MAFFT (Rozewicki et al. Citation2019) and removing gaps with Gblocks (Castresana Citation2000), a Maximum Likelihood (ML) phylogenetic tree was built with IQ-TREE (Nguyen et al. Citation2015) with bootstrap 1000 repetitions and the TVM + F + I + G4 model was chosen (). In the tree, P. quassioides was the sister group of Eurycoma longifolia, Leitneria floridana, and Ailanthus latissimus with high bootstrap support (100%). This result supports the fact that all those four species belong to the Simaroubaceae family.
Figure 1. Maximum-likelihood phylogenetic tree based on all protein-coding genes in the cp genomes of 23 Sapindales species. Bootstrap values (1000 replicates) are shown at the nodes.
Ethics approval and consent to participate
The data collection of plants was carried out with permission of related institution, and complied with national or international guidelines and legislation.
Author contributions
R.C. involved in the conception and design; Q.L., X.G., H.H., and Y.G. performed data analysis and interpretation; Q.L., H.Z., H.H, and Y.Z. drafted the manuscript; Q.L. and R.C reviewed the manuscript. All authors have read and agreed to the final approval of the version to be published. All authors have agreed to be accountable for all aspects of the work.
Disclosure statement
No potential conflict of interest was reported by the authors.
Data availability statement
The data that support the findings of this study are openly available in GenBank of NCBI at https://www.ncbi.nlm.nih.gov, under the accession number MZ902043. The associated BioProject, SRA, and BioSample numbers are PRJNA762200, SRR15839410, and SAMN21380154, respectively.
Additional information
Funding
References
- Alves IABS, Miranda HM, Soares LAL, Randau KP. 2014. Simaroubaceae family: botany, chemical composition and biological activities. Rev Bras Farmacogn-Braz J Pharmacogn. 24(4):481–501.
- Bai M, Zhao WY, Xu W, Zhang YY, Huang XX, Song SJ. 2020. Triterpenoids from Picrasma quassioides with their cytotoxic activities. Phytochem Lett. 39:128–131.
- Bolger AM, Lohse M, Usadel B. 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 30(15):2114–2120.
- Castresana J. 2000. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol. 17(4):540–552.
- Jin JJ, Yu WB, Yang JB, Song Y, dePamphilis CW, Yi TS, Li DZ. 2020. GetOrganelle: a fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 21(1):241.
- Nguyen LT, Schmidt HA, Von Haeseler A, Minh BQ. 2015. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. 32(1):268–274.
- Rozewicki J, Li S, Amada KM, Standley DM, Katoh K. 2019. MAFFT-DASH: integrated protein sequence and structural alignment. Nucleic Acids Res. 47(W1):W5–W10.
- Shi L, Chen H, Jiang M, Wang L, Wu X, Huang L, Liu C. 2019. CPGAVAS2, an integrated plastome sequence annotator and analyzer. Nucleic Acids Res. 47(W1):W65–W73.