
Abstract
We report the complete mitochondrial genome of Leocrates chinensis Kinberg, 1866 – the type species of the genus. It is 15061 bp long, and contains 13 protein-coding genes (PCGs), 22 tRNA genes (tRNAs), and 2 rRNA genes (rRNAs), and 1 putative control region. Phylogenetic analysis indicated that L. chinensis was placed as sister to Sirsoe methanicola (BS = 100) of the same family Hesionidae.
Introduction
Leocrates chinensis Kinberg et al. Citation1866 is a hesionid widely distributed in southeastern Chinese waters. The holotype of L. chinensis was collected from Victoria Harbor, Hong Kong during the Swedish naval frigate Eugenie’s around-the-world cruise during 1851–1853, and was later briefly described by Kinberg et al. (Citation1866) as the type species of the newly established hesionid genus Leocrates. A recent study redescribed L. chinensis with morphological details based on specimens from the type locality (Wang et al. Citation2018). Nevertheless, since the specimens were preserved in formalin, no attempt was made to extract DNA and determine the placements of this species and Hesionidae in tree of life. Therefore, we conducted genome skimming (Zhang et al. Citation2018) of Leocrates chinensis to help clarify the species status of Leocrates specimens that have been identified as L. chinensis based on cox1 (accession number: OL763897) and several other loci (i.e. 16S, 18S, and 28S, with accession numbers: OL764386, OP104345, and OP104348, respectively) that have been used in species classification of Hesionidae (Rouse et al. Citation2018; Wang et al. Citation2020), and to provide resources for studies of mitochondrial genome evolution in the family that currently has only one sequenced mitochondrial genome (Sirsoe methanicola, accession no.: OM914591; Lim et al. Citation2022).
Materials
The single Leocrates chinensis specimen used in this study was collected from the subtidal muddy sediment (Site M(4), 22°14.160′N, 114°11.115′E, water depth 9.1 m) of the Hong Kong, China on 10 June 2020. The specimen was fixed and preserved in 100% ethanol, and both the specimen and its genomic DNA are now deposited in the Biology of Marine Benthic Invertebrates Group, College of Ocean and Earth Sciences, Xiamen University (https://www.xmu.edu.cn/, Zhi Wang, [email protected]) under the voucher number XMU-Pol-2021-354 ().
Figure 1. Species reference image of Leocrates chinensis (voucher no: XMU-Pol-2021-354). This specimen was collected from subtidal muddy sediment (22°14.160′N, 114°11.115′E, water depth 9.1 m) of the Hong Kong, China on 10 June 2020, and is now deposited in the Biology of Marine Benthic Invertebrates Group, Xiamen University (Photo: Zhi Wang).

Methods
Whole genomic DNA was extracted by using TIANamp Genomic DNA Kit (TIANGEN, Beijing, China). The sequencing library was produced by using the Illumina Truseq™ DNA Sample Preparation Kit (Illumina, San Diego, USA) according to the manufacturer’s recommendations. The prepared library was loaded on the Illumina Novaseq 6000 platform for PE 2 × 150 bp sequencing at Novogene Company (Beijing, China). Sequence quality of raw genomic data was assessed using FastQC v.0.11.5 software (http://www.bioinformatics.babraham.ac.uk/projects/fastqc). Quality trimming and filtering of data was performed using fastp v.0.23.2 (Chen et al. Citation2018), reads containing more than 5% unknown nucleotides, and low-quality reads (reads containing more than 50% bases with Q-value ≤20) and all unpaired reads were discarded. To ensure the consistency in assembly result, the genome assembly was conducted using two methods, i.e. the filtered data were used to assemble the complete mitochondrial genome using the GetOrganelle v.1.7.6.1 pipeline with ‘animal_mt’ in the default database as seed reads (Jin et al. Citation2020) and NovoPlasty v.4.3.1 with the cox1 (OL763897) sequence as the seed reads (Dierckxsens et al. Citation2017). To check the coverage depth of the assembled mitogenome, “samtools depth” command in Samtools v.1.6 (Danecek et al. Citation2021) was used to calculate the coverage depth, and Circos v.0.69 (Krzywinski et al. Citation2009) was used to draw the coverage plot (as shown in ). The annotation of the mitogenome was made with the MITOS2 webserver (Donath et al. Citation2019) and the Mitoz annotation module (Meng et al. Citation2019). Additionally, we used maximum-likehood method with the software Phylosuite to reconstruct the phylogenetic tree (Zhang et al. Citation2020) among Errantia polychaete families. For the three Nereidiformia families, i.e. Hesionidae, Syllidae, and Nereididae, sequences of two genera in each family were included in the analyses; and for the other families including the outgroups (i.e. Capitellidae, Urechidae, Lumbricidae), only one genera was used for each family. All the mitogenomic data included in the analysis came from those published papers (), and concatenated protein-coding genes (PCGs) of these mitogemones were used in the analysis with each coding gene aligned individually using Mafft v.7.313 (Katoh and Standley Citation2013) under Codon alignment mode. Ambiguously aligned regions were removed with Gblocks v.0.91 with default settings (Castresana Citation2000). The best-fitting partition models (GTR + F + I + G4) for ML analysis were selected by the software ModelFinder (Kalyaanamoorthy et al. Citation2017).
Figure 2. The complete mitogenome of Leocrates chinensis. The middle circles and innermost represent depth distribution and GC content, respectively. The outermost circle shows gene arrangements, with green for PCGs fragments, orange for rRNAs and red for tRNAs.
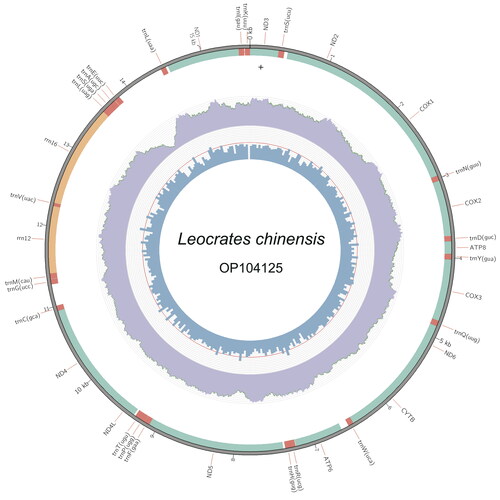
Table 1. Mitochondrial sequences used in the phylogenetic analysis as shown in .
Results
The two assembly pipelines (i.e. GetOrganelle and NovoPlasty) gave an exact identical circular mitogenome. The size of the complete mitochondrial genome was 15,061 bp, and the data were submitted to the NCBI (accession no.: OP104125). The genome consisted of 60.3% A + T, with 28.8% of A, 31.5% of T, 14.8% of G, and 24.9% of C. The genome contains 13 PCGs, i.e. 2 rRNA genes, 22 tRNA genes, and 1 putative control region consisting of 620 bp (). Six PCGs started with ATG codon, two PCGs started with ATT codon, one PCG started with ATC and four PCGs started with ATA. All 13 PCGs terminated with TAA stop codon except nad4 and nad2, which with T stop codon. The real phylogenetic relationships among available Errantia polychaetous families are exhibited in . Results showed that L. chinensis was sister taxon of Sirsoe methanicola (BS = 100), which was the only one sequenced mitochondrial genome in the same family Hesionidae.
Figure 3. Phylogenetic tree reconstructed with Maximum Likelihood (ML) method. The tree was constructed based on concatenated nucleotide sequences of 13 protein-coding genes (PCGs) of 20 polychaetous species in 17 families and 1 clitellate species. Values of robustness were calculated from ML analyses, and only bootstrap (BS) values ≥50 are shown at nodes. GenBank accession numbers used are listed after the species names. The scale bar indicates the number of substitutions per site.

Discussion and conclusion
The first complete mitochondrial genome of the polychaetous species Leocrates chinensis is reported in this study, which is also the first mitogenome reported in the genus Leocrates. We used two different assembly methods (i.e. GetOrganelle v.1.7.6.1 and NovoPlasty v.4.3.1) to ensure the consistency of the assembly result. We noted that there was slight negative coverage anomaly in the control region (CR). It might be due to compositional bias affecting sequencing. As we all know, the control region has high A + T content, and this region always facing sequencing problems. Based on our experiences in mitogenomic studies of polychaetes, the CR is difficult to assemble and it always has a slight negative coverage anomaly. This study provides a valuable data to studying the phylogenetic and evolutionary history of annelid polychaetes. And to make better understanding of the phylogeny within Hesionidae, mitogenomic studies on more genera of this family, such as Oxydromus, Podarkeopsis, Micropodarke, etc., should be put into agenda in future studies.
Ethical approval
This work using animal species not protected by regulations or law. An ethical review by the Statement Animal Experiment Committee was therefore not required.
Author contributions
ZW, HZ, LG, and CK initiated and designed this project. ZW and CK jointly supervised the work. JWQ collected the samples. JWQ and ZW conducted the morphological identification. DY, and SL analyzed the data. XL, PL, DM, ZW, DY, and JWQ jointly drafted the manuscript text. All authors contributed to the review, critical revision and preparation of the manuscript, and the final approval of the version to be published, and that all authors agree to be accountable for all aspects of the work.
Acknowledgements
The authors thank Vicky Chu from Hong Kong Baptist University and Yanjie Zhang from School of Life Sciences, Hainan University for preparing and delivering the specimens.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
The genome sequence data that support the findings of this study are openly available in GenBank of NCBI at [https://www.ncbi.nlm.nih.gov](https://www.ncbi.nlm.nih.gov/) under the accession no. OP104125. The associated BioProject, Bio-Sample, and SRA numbers are PRJNA859499, SAMN29785536, and SRR20276768, respectively.
Additional information
Funding
References
- Aguado MT, Glasby CJ, Schroeder PC, Weigert A, Bleidorn C. 2015. The making of a branching annelid: an analysis of complete mitochondrial genome and ribosomal data of Ramisyllis multicaudata. Sci Rep. 5(1):12072–12013.
- Alves PR, Halanych KM, Santos CSG. 2020. The phylogeny of Nereididae (Annelida) based on mitochondrial genomes. Zoologica Scripta. 49(3):366–378.
- Bleidorn C, Podsiadlowski L, Bartolomaeus T. 2006. The complete mitochondrial genome of the orbiniid polychaete Orbinia latreillii (Annelida, Orbiniidae)–a novel gene order for Annelida and implications for annelid phylogeny. Gene. 370:96–103.
- Boore JL, Brown WM. 2000. Mitochondrial genomes of Galathealinum, Helobdella, and Platynereis: sequence and gene arrangement comparisons indicate that Pogonophora is not a phylum and Annelida and Arthropoda are not sister taxa. Mol Biol Evol. 17(1):87–106.
- Castresana J. 2000. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol. 17(4):540–552.
- Chen X, Li M, Liu H, Li B, Guo L, Meng Z, Lin H. 2016. The complete mitochondrial genome of the polychaete, Goniada japonica (Phyllodocida, Goniadidae). Mitochondrial DNA A DNA Mapp Seq Anal. 27(4):2850–2851.
- Chen S, Zhou Y, Chen Y, Gu J. 2018. FASTP: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics. 34(17):i884–i890.
- Chen X, Yang W, Si Y, Li B, Xu R, et al. 2019. The complete mitochondrial genome of the polychaete, Marphysa tamurai (Eunicida, Eunicidae). Mitochondrial DNA Part B. 4(1):1055–1056.
- Danecek P, Bonfield JK, Liddle J, Marshall J, Ohan V, Pollard MO, Whitwham A, Keane T, McCarthy SA, Davies RM, Li H. 2021. Twelve years of SAMtools and BCFtools. Gigascience. 10(2):giab008.
- Dierckxsens N, Mardulyn P, Smits G. 2017. NOVOPlasty: de novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 45(4):e18.
- Donath A, Jühling F, Al-Arab M, Bernhart SH, Reinhardt F, Stadler PF, Middendorf M, Bernt M. 2019. Improved annotation of protein-coding genes boundaries in metazoan mitochondrial genomes. Nucleic Acids Res. 47(20):10543–10552.
- Hamilton LE, Preston GC, Wheeler JB, Janosik AM, Bogantes VE. 2022. The complete mitochondrial genome of the plumed worm Diopatra cuprea (Annelida: onuphidae). Mitochondrial DNA B Resour. 7(1):49–50.
- Jin J-J, Yu W-B, Yang J-B, Song Y, dePamphilis CW, Yi T-S, Li D-Z. 2020. GetOrganelle: a fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 21(1):241.
- Kalyaanamoorthy S, Minh BQ, Wong TKF, von Haeseler A, Jermiin LS. 2017. ModelFinder: fast model selection for accurate phylogenetic estimates. Nat Methods. 14(6):587–589.
- Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 30(4):772–780.
- Kinberg JGH. 1866. Annulata nova (Nephtydea Phyllodocea Alciopea Hesionida Glycerea Goniadea Syllidea Ariciea Spiodea Aonidea Cirratulida, Opheliacea (1865). Öfv Kong Vetensk-Akad Förhand. 22(239):258.
- Kobayashi G, Itoh H, Nakajima N. 2022. First mitochondrial genomes of Capitellidae and Opheliidae (Annelida) and their phylogenetic placement. Mitochondrial DNA B Resour. 7(4):577–579.
- Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R, Horsman D, Jones SJ, Marra MA. 2009. Circos: an Information Aesthetic for Comparative Genomics. Genome Res. 19(9):1639–1645.
- Lim SJ, Thompson LR, Young CM, Gaasterland T, Goodwin KD. 2022. Dominance of Sulfurospirillum in metagenomes associated with the methane ice worm (Sirsoe methanicola). Appl Environ Microbiol. 88(15):e00290-22.
- Meng G, Li Y, Yang C, Liu S. 2019. MitoZ: a toolkit for animal mitochondrial genome assembly, annotation and visualization. Nucleic Acids Res. 47(11):e63.
- Richter S, Schwarz F, Hering L, Böggemann M, Bleidorn C. 2015. The utility of genome skimming for phylogenomic analyses as demonstrated for glycerid relationships (Annelida, Glyceridae). Genome Biol Evol. 7(12):3443–3462.
- Rouse GW, Carvajal JI, Pleijel F. 2018. Phylogeny of Hesionidae (Aciculata, Annelida), with four new species from deep-sea eastern Pacific methane seeps, and resolution of the affinity of Hesiolyra. Invertebrate Systemat. 32:1050–1068.
- Vallès Y, Halanych KM, Boore JL. 2008. Group II introns break new boundaries: presence in a bilaterian’s genome. PLOS One. 3(1):e1488.
- Wang Z, Qiu J-W, Salazar-Vallejo SI. 2018. Redescription of Leocrates chinensis Kinberg, 1866 (Annelida, Hesionidae). Zoolog Stud. 57(5):1–11.
- Wang Z, Li X, Xu Z, Liu H, Wang Y, Zheng F. 2019. First report of the complete mitochondrial genome and phylogenetic analysis of Aphrodita australis (Aphroditidae, Annelida). Mitochondrial DNA B Resour. 4(2):4116–4117.
- Wang Z, Xu T, Zhang Y, Zhou Y, Liu Z, Chen C, Watanabe HK, Qiu J-W. 2020. Molecular phylogenetic and morphological analyses of the ‘monospecific’ Hesiolyra (Annelida: Hesionidae) reveal two new species. Deep Sea Res Part I Oceanogr Res Papers. 166:103401.
- Weigert A, Golombek A, Gerth M, Schwarz F, Struck TH, Bleidorn C. 2016. Evolution of mitochondrial gene order in Annelida. Mol Phylogenet Evol. 94(Pt A):196–206.
- Wu Z, Shen X, Sun M, Ren J, Wang Y, Huang Y, Liu B. 2009. Phylogenetic analyses of complete mitochondrial genome of Urechis unicinctus (Echiura) support that Echiurans are derived annelids. Mol Phylogenet Evol. 52(2):558–562
- Zhang D, Gao F, Jakovlić I, Zou H, Zhang J, Li WX, Wang GT. 2020. PhyloSuite: an integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol Ecol Resour. 20(1):348–355.
- Zhang Q, Liu H, Zhang Y, Ruan H. 2019. The complete mitochondrial genome of Lumbricus rubellus (Oligochaeta, Lumbricidae) and its phylogenetic analysis. Mitochondrial DNA B Resour. 4(2):2677–2678.
- Zhang Y, Sun J, Rouse GW, Wiklund H, Pleijel F, Watanabe HK, Chen C, Qian P-Y, Qiu J-W.,. 2018. Phylogeny, evolution and mitochondrial gene order rearrangement in scale worms (Aphroditiformia, Annelida). Mol Phylogenet Evol. 125:220–231.