
Abstract
Juglans mandshurica Maxim., 1856 is a second-class, protected, rare tree species of high economic and ecological value. We elucidated the complete mitochondrial (mt) genome of J. mandshurica using the Illumina Novaseq 6000 and Nanopore platforms. The complete sequences of 558,032 and 161,386 bp had an overall GC content of 45.0% and 45.3%, respectively, and 61 genes could be annotated, including 38 protein-coding, 20 tRNA, and 3 rRNA genes. The high-quality J. mandshurica mt genomic sequences presented in this study will serve as a useful resource for a range of genetic, functional, evolutionary, and comparative genomic studies on this species of the Juglandaceae family.
Introduction
Juglans mandshurica Maxim. is a second-class, protected, rare tree species that belongs to the genus Juglans of the Juglandaceae family (Wang et al. Citation2010). It is primarily scattered in mixed forest regions of northeastern China, northern Korea, and far eastern Russia, at altitudes of 400–1000 m (Chen et al. Citation2018; Liu et al. 2020). J. mandshurica has high ecological and economic value and is widely used as a source of wood, food, and medicine (Huang et al. Citation2010; Shen et al. Citation2015; Luo et al. Citation2017). Notably, it is a straight-root tree with strong tolerance to cold and drought that can survive at temperatures as low as −50 °C. Consequently, it is often employed as the parent plant for breeding cold-resistant Juglans regia (Liu Citation2020).
Plant mitochondrial (mt) genomes tend to have a more heterogeneous composition than chloroplast (cp) genomes (Zhang et al. Citation2019). Owing to the conservation of their genomic structure and a fairly low recombination rate, cp genomes provide valuable information for taxonomic classification and phylogenetic reconstruction, which aid in the identification of complex evolutionary relationships (Jansen et al. Citation2008; Parks et al. Citation2009; Zhang et al. Citation2019). In contrast, it is well recognized that the highly dynamic mt genome structure can have a variety of genomic conformations owing to recombination and interference based on repeated content (Palmer and Herbon Citation1988; Marechal and Brisson Citation2010). These characteristics make the plant mt genome a fascinating study topic, and as of March 24, 2022, as many as 437 plant mt sequences have been published (https://www.ncbi.nlm.nih.gov/genome/browse#!/organelles/). Although many studies have concentrated on the ecological characteristics, cultivation techniques, and chemical composition of J. mandshurica, there are very few exploring its genome (Wang et al. Citation2018; Pang, Citation2021; Liu et al. Citation2021). To our knowledge, the present study is the first report of the J. mandshurica mt genome.
Materials and methods
Plant materials and sequencing
Fresh J. mandshurica leaves were collected in July 2021 from the Forestry Experimental Station (37.52°N, 121.34°E) at Ludong University, Yantai City, China. Plant specimens (barcode number SDF1000958) and total genomic DNA (code number htq2021cp10) were stored at the Shandong Provincial Center of Forest and Grass Germplasm Resources (Biao Han, [email protected]). Total DNA was obtained using the Blood/Cell/Tissue Genomic DNA Extraction Kit (TIANamp Genoic DNA Kit, Tiangen, Beijing, China) (Uddin et al. Citation2014). The DNA library was constructed using the Ligation Sequencing Kit (SQK-LSK109) and sequenced using the Nanopore PromethION sequencing platform (Nanodrop Technologies, Wilmington, DE, US); 11.54 Gb raw data with an N50 length of 13,783 bp was generated. The data was filtered and re-edited using NanoFilt and NanoPlot in the Nanopack software (De Coster et al. Citation2018), resulting in 10.71 G clean reads. Libraries with an average fragment length of 350 bp were constructed; they were sequenced using the Illumina Novaseq 6000 platform (Illumina, San Diego, CA, USA) and 8.83 Gb raw data was generated. After editing the data using the NGS QC ToolKit v2.3.3 software (Patel and Jain, Citation2012), 8.78 Gb of data was generated from 29.28 million reads. The read coverage depth of Illumina and Nanopore sequencing exceeded 100×, indicating good sequencing data results (Figure S1).
Figure 1. Morphological characteristics of Juglans mandshurica. (a) whole plant, (b) fruits, (c) inflorescence, (d) seeds, (e) leaves.

Genome assembly and annotation
We first obtained a rough but computationally efficient assembly using the Miniasm tool (Li, Citation2016) after trimming adapter sequences with the Porechop software (https://github.com/rrwick/Porechop) and polished the resulting assembly with the Racon tool (https://github.com/isovic/racon). We selected contigs with homology to Fagus sylvatica (NCBI Reference Sequence: MT446430.1) mt genome using the Bandage software (Wick et al. Citation2015), retaining contigs with at least one ≥5 kb alignment to the F. sylvatica mitochondrion using the BLASTn tool (Chen et al. Citation2015). We then proceeded to align the Nanopore reads to our draft F. sylvatica assembly with the minimap2 tool (Li, Citation2018), segregated aligned reads, and assembled them de novo first using the Unicycler software (Wick et al. Citation2017) and then with the Flye software (Kolmogorov et al. Citation2019), as described previously. The final genome sequence was obtained by polishing with the Pilon tool using Illumina Novaseq 6000 sequencing reads. The mt genomes were annotated using the BLASTn, MITOFY (Alverson et al. Citation2010), and MFannot tools (http://megasun.bch.umontreal.ca/cgi-bin/mfannot/) and angiosperm mt genes were used to query sequences in the NCBI database (https://www.blast.ncbi.nlm.nih.gov). The tRNA genes were identified using the tRNA scan-SE software (http://lowelab.ucsc.edu/tRNAscan-SE/). The genome circle map of mt genome was visualized with the OGDRAW v1.2 software (http://ogdraw. mpimp-golm.mpg.de/). Manual correction was performed after software annotation to ensure the accuracy of annotation.
Phylogenetic analysis
The phylogeny of J. mandshurica was analyzed by aligning the mt genomic sequences of 18 species using the HomBlocks software (Bi et al. Citation2018) and subsequent trimming using the Gblock method. The Model-finder software was employed to chop the model WAG + I + G, followed by construction of the maximum likelihood (ML) phylogenetic tree using the RAxML v8.2.9 software with 1,000 bootstrap replicates (Stamatakis, Citation2014).
Results
Morphological characteristics
We obtained many photographs while observing and recording the morphological characteristics of J. mandshurica, including photographs of the whole plant, fruits, inflorescence, seeds, and leaves (). J. mandshurica is an arbor with imparipinnate leaves. The fruits are globose, ovate, or elliptic. Flowering occurs in May, and the fruit stage is in August–September.
Genome sequencing assembly and characterization
Ample evidence suggests that plant mt genomes can exist in multiple circular configurations and even noncircular forms owing to intramolecular recombination events mediated by repeats (Kozik et al. Citation2019; Zhang et al. Citation2019; Logacheva et al. Citation2020). We obtained two complete circular J. mandshurica mt genomic molecules (558,032 bp and 161,386 bp) from the de novo assembly of the filtered mt reads with overall GC contents of 45.0% and 45.3%, respectively. Circular genome maps were constructed using the OGDRAW software (), and 61 total genes, including 38 protein-coding genes, 20 tRNA genes, and 3 rRNA genes, were subsequently annotated (). Among these, 38 protein-coding genes were found to contain exons: atp1, atp4, atp6, atp8, atp9, ccmB, ccmC, ccmFN, cob, cox1, cox3, matR, mttB, nad3, nad4L, nad6, nad9, rpl5, rpl10, rpl16, rps1, rps3, rps4, rps7, rps10, rps12, rps14, rps19, sdh3, and sdh4 with one exon; ccmFc, cox2, and rpl2 with two exons; nad4 with four exons; and nad1, nad2, nad5, and nad7 with five exons. Moreover, eight protein-coding genes were found to contain introns: ccmFc, cox2, and rpl2 with one intron; nad4 with three introns; and nad1, nad2, nad5, and nad7 with four introns. Notably, nad1, nad2, and nad5 were found to undergo trans-splicing.
Figure 2. J. mandshurica mitochondrial (mt) genome gene map. The gene map denotes annotated genes based on different functional groups, which are color-coded on the outer circle as transcribed clock-wise (outside) and counter clock-wise (inside). The inner circle indicates the GC content as a dark grey plot.
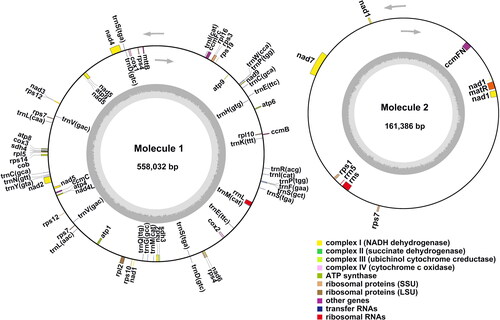
Table 1. Gene composition of the J. mandshurica mt genome.
Phylogenetic analysis
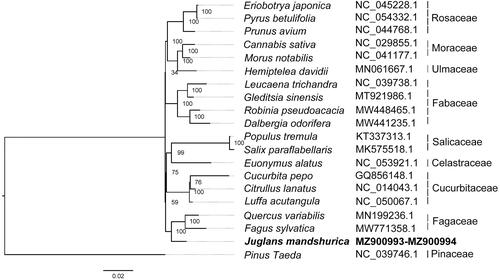
Phylogenetic analysis was performed using the ML method, based on the mt genome sequences of 19 species: including Eriobotrya japonica, Pyrus betulifolia, Prunus avium, Cannabis sativa, Morus notabilis, Hemiptelea davidii, Leucaena trichandra, Gleditsia sinensis, Robinia pseudoacacia, Dalbergia odorifera, Populus tremula, Salix paraflabellaris, Euonymus alatus, Cucurbita pepo, Citrullus lanatus, Luffa acutangula, Quercus variabilis, F. sylvatica, and Pinus Taeda. Our results revealed that J. mandshurica is most closely related to Q. variabilis and F. sylvatica ().
Figure 3. Maximum likelihood (ML) phylogenetic tree of J. mandshurica and 19 other species based on the conserved segments of mitogenome sequences, with Pinus taeda as the outgroup. The accession numbers are showed in the figure, and the numbers behind each node are bootstrap support values.
Discussion and conclusion
Juglans L. is an economically and ecologically important genus of the Juglandaceae family (Hu et al. Citation2016). A total of five species (J. regia, J. sigillata, J. cathayensis, J. hopeiensis, and J. mandshurica) and one variety (J. draconia) belonging to this genus are found in China. The Juglans L. cp genome is highly conserved, with a typical four-segmented structure (LSC, SSC, IRa, IRb) (She, Citation2021). In contrast, mt genomes are highly diverse, which is evident based on the different plant mitogenome sequences that have been reported (Kozik et al. Citation2019; Liu et al. Citation2022). Although plant mitogenomes are often assembled and displayed as circular maps, they mostly exist as complex and dynamic collections of linear DNA interspersed with smaller circular and branched configurations (Oldenburg and Bendich et al. Citation2015; Morley and Nielsen Citation2017; Wynn and Christensen Citation2018; Kozik et al. Citation2019). For example, the Quercus acutissima mt genome is a branched structure comprising one linear and two circular formations (Liu et al. Citation2022), the Abelmoschus esculentus mt genome is composed of one linear and one circular structure (Li et al. Citation2022), and the Ipomoea batatas mt genome is arranged as four circular sections (Yang et al. Citation2022). The present study demonstrates that the J. mandshurica mt genome comprises two circular formations (558,032 bp and 161,386 bp); however, the coexistence of these molecules requires further investigation.
Given the low substitution rate in the mt genome, the constituent genes are a valuable source of information for phylogenetic analysis at high taxonomic levels (Hiesel et al. Citation1994). Our analysis of the phylogenetic relationships of J. mandshurica based on its mitochondrial genomic information revealed its close relationship with Q. variabilis and F. sylvatica. The high-quality mt genomic sequences presented in this study will serve as an important resource for a range of genetic, functional, evolutionary, and comparative genomic studies on J. mandshurica and other Juglans species of the Juglandaceae family.
Ethical approval
The authors complied with the international Union for Conservation of Nature (IUCN) policies research involving species at risk of extinction (see Guidelines for appropriate uses of IUCN Red List data), the Convention on Biological Diversity and the Convention on Trade in Endangered Species of Wild Fauna and Flora.
Author contributions
Xun Su (Data analysis and Thesis writing); Qiong Liu (Data analysis and Paper revision); Haili Guo (Data analysis and Thesis writing); Dechang Hu (Experimental design); Dan Liu (Experimental design and Paper revision); Zihao Wang (Resource investigation and Data interpretation); Ping Zhang (Data interpretation), and that all authors agree to be accountable for all aspects of the work.
Supplemental Material
Download MS Word (73.5 KB)Disclosure statement
The authors declare no conflict of interest.
Data availability statement
The genome sequence data are openly available in GenBank of NCBI at (https://www.ncbi.nlm.nih.gov/) under the accession no. MZ900993 and MZ900994. The associated BioProject, SRA, and Bio-Sample numbers are PRJNA757787, SRR15615977, SRR15615978, and SAMN20981794 respectively.
Additional information
Funding
References
- Alverson AJ, Wei XX, Rice DW, Stern DB, Barry K, Palmer JD. 2010. Insights into the evolution of mitochondrial genome size from complete sequences of Citrullus lanatus and Cucurbita pepo (Cucurbitaceae). Mol Biol Evol. 27(6):1436–1448.
- Bi G, Mao Y, Xing Q, Cao M. 2018. HomBlocks: a multiple-alignment construction pipeline for organelle phylogenomics based on locally collinear block searching. Genomics. 110(1):18–22.
- Chen Y, Ye W, Zhang Y, Xu Y. 2015. High speed BLASTN: an accelerated MegaBLAST search tool. Nucleic Acids Res. 43(16):7762–7768.
- Chen DP, You JH, Liu DD, Wang XY, Li Y, Xu GZ, Lu JM. 2018. A comparative study on evolutionary structures of the Juglans mandshurica different ecological environment stress. J. Northeast Univ Nat Sci Ed. 50(02):97–102.
- De Coster W, D'Hert S, Schultz DT, Cruts M, Van Broeckhoven C. 2018. NanoPack: visualizing and processing long read sequencing data. Bioinformatics. 34(15):2666–2669.
- Hiesel R, von Haeseler A, Brennicke A. 1994. Plant mitochondrial nucleic acid sequences as a tool for phylogenetic analysis. Proc Natl Acad Sci U S A. 91(2):634–638.
- Hu YH, Woeste KE, Zhao P. 2016. Completion of the chloroplast genomes of five chinese juglans and their contribution to chloroplast phylogeny. Front Plant Sci. 7:1955.
- Huang GL, Mao LR, Liu YF, Qin DZ, Sun L, Zhang HJ. 2010. Economic value and cultivation techniques of Juglans mandshurica. Forest Sci Technol.. (3):52–53,56.
- Jansen RK, Cai Z, Raubeson LA, Daniell H, Boore JL. 2008. Analysis of 81 genes from 64 plastid genomes resolves relationships in angiosperms and identifies genome-scale evolutionary patterns. Proc Natl Acad Sci U S A. 104(49):19369–19374.
- Kolmogorov M, Yuan J, Lin Y, Pevzner PA. 2019. Assembly of long, error-prone reads using repeat graphs. Nat Biotechnol. 37(5):540–546.
- Kozik A, Rowan BA, Lavelle D, Berke L, Schranz ME, Michelmore RW, Christensen AC. 2019. The alternative reality of plant mitochondrial DNA: one ring does not rule them all. Plos Genet. 15(8):e1008373.
- Li H. 2016. Minimap and miniasm: fast mapping and de novo assembly fornoisy long sequences. Bioinformatics. 32(14):2103–2110.
- Li H. 2018. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics. 34(18):3094–3100.
- Li JN, Gao RX. 2020. Research progress on the genetic breeding of Juglans mandshurica in China. J Anhui Agri Sci. 48(17):4–7.
- Li J, Li J, Ma Y, Kou L, Wei J, Wang W. 2022. The complete mitochondrial genome of okra (Abelmoschus esculentus): using nanopore long reads to investigate gene transfer from chloroplast genomes and rearrangements of mitochondrial DNA molecules. BMC Genom. 23:481.
- Liu DN. 2020. Effects of different seedling rearing methods and fertilization on growth and physiology of Juglans mandshurica Maxim. Mudanjiang Normal University. China (master thesis).
- Liu D, Guo H, Zhu J, Qu K, Chen Y, Guo Y, Ding P, Yang H, Xu T, Jing Q, et al. 2022. Complex physical structure of complete mitochondrial genome of quercus acutissima (fagaceae): a significant energy plant. Genes. 13(8):1321.
- Liu H, Song Q, Wang TM, Zhang H, Zhai YJ, Kang YG. 2021. Establishment of content determination of 5 components and their content difference analysis in the branch and root of Juglans mandshurica. China Pharm. 32(08):933–939.
- Logacheva MD, Schelkunov MI, Fesenko AN, Kasianov AS, Penin AA. 2020. Mitochondrial genome of Fagopyrum esculentum and the genetic diversity of extranuclear genomes in buckwheat. Plants. 9(5):618.
- Luo M, Qiao Q, Mu YS, Ruan X, Yang X, Xing WM, Duan XH. 2017. Temporal and spatial variations of active ingredients in Juglans mandshurica Maxim and antitumor their anti-tumor activity. Bull Bot Res. 37(6):947–952.
- Marechal A, Brisson N. 2010. Recombination and the maintenance of plant organelle genome stability. New Phytol. 186(2):299–317.
- Morley SA, Nielsen BL. 2017. Plant mitochondrial DNA. Front Biosci. 22(6):1023–1032.
- Oldenburg DJ, Bendich AJ. 2015. DNA maintenance in plastids and mitochondria of plants. Front Plant Sci. 6:883.
- Palmer JD, Herbon LA. 1988. Plant mitochondrial DNA evolves rapidly in atructure, but alowly in Herbon sequence. J Mol Evol. 28(1-2):87–97.
- Pang Y. 2021. Exploration of the cultivation technology of Juglans mandshurica. Seed Sci Technol. 39(22):113–114.
- Parks M, Cronn R, Liston A. 2009. Increasing phylogenetic resolution at low taxonomic levels using massively parallel sequencing of chloroplast genomes. BMC Biol. 7:84.
- Patel RK, Jain M. 2012. NGS QC Toolkit: a toolkit for quality control of next generation sequencing data. PLoS One. 7(2):e30619.
- She R. 2021. Population genomics and phylogeny of walnut (Juglans). Northwest University. China (master thesis).
- Shen GZ, Zou GH, Liang T, Liang QC, Wu YY. 2015. Chemical constituents of Juglans mandshurica. Chin J Exp Trad Med Form. 21(17):219–224.
- Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 30(9):1312–1313.
- Uddin MS, Sun W, He X, Teixeira Da Silva JA, Cheng QX. 2014. An improved method to extract DNA from mango Mangifera indica. Biologia. 69(2):133–138.
- Wang DN, Mu CC, Feng FJ. 2010. Establishment and optimization of ISSR-PCR reaction system for Juglans mandshurica Maxim. Res Exp Lab. 29(11):18–22.
- Wang TM, Xu SZ, Yu WJ, Di X, Zhang H, Zhai YJ, Kang YG. 2018. Morphological study of Juglans mandshurica. Chin Arch Trad Chin Me. 36(03):675–677.
- Wick RR, Schultz MB, Zobel J, Holt KE. 2015. Bandage: interactive visualization of de novo genome assemblies. Bioinformatics. 31(20):3350–3352.
- Wick RR, Judd LM, Gorrie CL, Holt KE. 2017. Unicycler: resolving bacterial genome assemblies from short and long sequencing reads. PLOS Comput Biol. 13(6):e1005595.
- Wynn EL, Christensen AC. 2018. Repeats of unusual size in plant mitochondrial genomes: identification, incidence and evolution. G3-Genes Genomes Genet. 9(2):549–559.
- Yang Z, Ni Y, Lin Z, Yang L, Chen G, Nijiati N, Hu Y, Chen X. 2022. De novo assembly of the complete mitochondrial genome of sweet potato (Ipomoea batatas [L.] Lam) revealed the existence of homologous conformations generated by the repeat-mediated recombination. BMC Plant Biol. 22(1):285.
- Zhang F, Li W, Gao CW, Zhang D, Gao LZ. 2019. Deciphering tea tree chloroplast and mitochondrial genomes of Camellia sinensis var. assamica. Sci Data. 6(1).