
Abstract
Camellia atrothea H.T. Chang, H.S. Wang & B.H. Chen 1983 is a native Camellia species in China, which has significant economic and breeding values. Here, we assembled and analyzed the complete chloroplast genome sequence of C. atrothea based on the whole genome sequencing data from Illumina NovaSeq6000 platform. Results show that the complete chloroplast genome of C. atrothea is 157,099 bp in length, and comprises a large single-copy (LSC, 86,643 bp) region, a small single-copy (SSC, 18,276 bp) region, and a pair of inverted repeats (IRs, 26,090 bp) region. Annotation of the C. atrothea chloroplast genome predicts a total of 133 genes, including 88 protein-coding genes, 8 ribosomal RNA genes, and 37 transfer RNA genes. The overall GC content accounts for 37.30% of the genome, which is clearly consistent with that of other previously published Camellia species from section Thea. Phylogenetic analyses of the whole chloroplast genome sequences of 14 Camellia species using Actinidia chinensis as outgroup reveals that C. atrothea has a close relationship with C. leptophylla.
Introduction
Camellia atrothea is an economically important Camellia species of section Thea from Theaceae family (Chang Citation1981). It is mainly distributed in Yunnan Province, southwest of China (Ming and Bartholomew Citation2007). As a wild related species of cultivated tea plants (C. sinensis), C. atrothea contains rich tea quality-related secondary metabolites and excellent resistance gene resources, which shows great economic and breeding values for tea germplasm innovation and production. However, due to the excessive exploitation of the young leaves of this species to process tea, the wild habitats of C. atrothea has been seriously damaged, which is in urgent need of scientific conservation toward sustainable utilization of this valuable Camellia plants (Huang Citation2016).
Chloroplast genome sequences are crucial genetic resources in revealing the origin and genetic relationship of plant species, and particularly have great significance in species identification, classification, and conservation (Daniell et al. Citation2016). In the past decades, the innovation of next generation sequencing technology has largely promoted the nuclear and organelle genome sequencing of hundreds of flowering plants. Of them, a total of 67 chloroplast genome from Camellia plants, such as C. sinensis, C. taliensis, and C. oleifera, have been released, which has accelerated the phylogenetic resolution and conservation of Camellia plants (Huang et al. Citation2014; Yang et al. Citation2013). Nevertheless, the chloroplast genome of C. atrothea has yet to be reported, which has restricted the genetic conservation of C. atrothea for its better utilization in the future.
In the present study, we deeply sequenced and assembled the chloroplast genome of C. atrothea using whole genome sequencing data from Illumina platform. Through comprehensive annotation and evolution analysis, we aim to provide insights into the genome characterization and evolution of this important Camellia plant, which would help genetic research and breeding progress of cultivated tea plants in the future.
Materials and methods
Plant material collection and DNA extraction
The mature leaves tissues of C. atrothea were collected from Pingbian County, Yunnan Province, China (103°42′E, 22°58′N) in October 23th, 2017. The specimens were deposited at the State Key Laboratory of Tea Plant Biology and Utilization, Anhui Agricultural University under specimen number LH#134 (https://tealab.ahau.edu.cn; Prof. Enhua Xia, [email protected]). After collection, the leaves were stored in liquid nitrogen and the genomic DNA was extracted using polysaccharides and polyphenolics-rich plants kit following the manufacturer’s instruction (TIANGEN, China).
Library construction and sequencing
The high-quality DNA was used to construct a 500 bp insert-size paired-end sequencing library following the Illumina’s instructions. The constructed library was sequenced by Illumina NovaSeq6000 platform. The raw sequencing data was preprocessed using SolexaQA (Cox et al. Citation2010) to remove adaptors, low-quality base (phred score <20), short read (length < 30 bp), and potential contaminations.
Sequence assembly and annotation
The high-quality whole genome sequencing reads of C. atrothea were first to aligned to the chloroplast genome sequences of 55 Camellia plants using BWA software with default parameters. The aligned reads were then extracted and regarded as chloroplast-derived sequences. The sequencing reads of C. atrothea chloroplast genome were then assembled using GetOrganelle (Jin et al. Citation2020). Annotation of the chloroplast genome of C. atrothea was performed using GeSeq (Tillich et al. Citation2017), followed by manual adjustments according to the homologous genes of cultivated tea plants C. sinensis (MW148820). The resulted GenBank annotation file was used to visualize chloroplast genome map by CPGView (http://www.1kmpg.cn/cpgview/). The simple sequence repeat of C. atrothea chloroplast genome was identified using MISA package (Beier et al. Citation2017), and their polymorphic status among 67 Camellia plants were investigated using CandiSSR package (Xia et al. Citation2015).
Phylogenetic tree construction
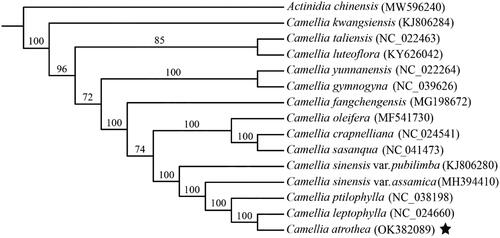
A total of 13 chloroplast genomes from Camellia species, including C. kwangsiensis (KJ806284), C. taliensis (NC_022463), C. luteoflora (KY626042), C. yunnanensis (NC_022264), C. gymnogyna (NC_039626), C. fangchengensis (MG198672), C. oleifera (MF541730), C. crapnelliana (NC_024541), C. sasanqua (NC_041473), C. sinensis var. pubilimba (KJ806280), C. sinensis var. assamica (MH394410), C. ptilophylla (NC_038198), C. leptophylla (NC_024660), and C. atrothea (OK382089), were downloaded from NCBI GenBank database. The whole chloroplast genomes of the 13 Camellia plants and outgroup Actinidia chinensis (MW596240) were then aligned using MAFFT with default parameter (Katoh and Standley Citation2013). The best suitable model for phylogeny construction was estimated by jModelTest (Darriba et al. Citation2012) from 88 different nucleotide substitution models using Bayesian Information Criterion. The phylogenetic tree was constructed using RAxML (Stamatakis Citation2014) package with default parameters. The bootstrap support value was calculated from 1000 iterations. The resulted tree was visualized using MEGA software (Tamura et al. Citation2013).
Results
We sequenced and analyzed the whole genome sequence of C. atrothea using Illumina NovaSeq6000 technology. This generates a total of 374,897,845 high-quality reads, of which 8,913,959 were chloroplast-derived reads and well aligned with the published chloroplast genome sequences of 13 Camellia plants from section Thea (). The average coverage of the chloroplast DNA was 8511×. The obtained chloroplast-derived clean reads were then assembled using GetOrganelle and further annotated using GeSeq by comparing with homologous genes of cultivated tea plants C. sinensis (MW148820; Chen et al. Citation2021). The complete chloroplast genome sequences of C. atrothea together with its annotations have been deposited into the NCBI GenBank database under the accession number of OK382089.
Figure 1. Sample collection of Camellia atrothea used for DNA sequencing in the present study. (A) Whole plant of the collected individual (imaged by Yanli Wang). (B) Mature leaves used for sequencing. (C) Statistics of the sequencing data.

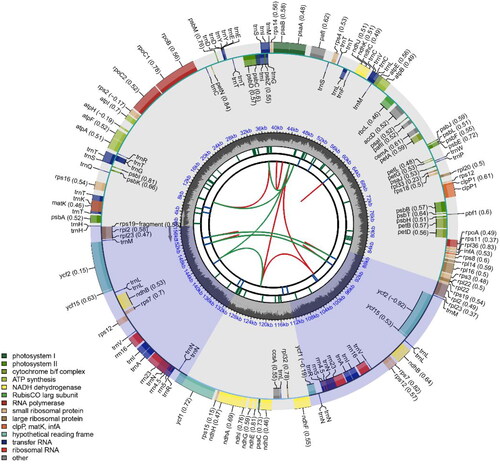
The chloroplast genome of C. atrothea is 157,099 bp in length, and structurally contains a small single-copy region (18,276 bp), a large single-copy region (86,643 bp), and a pair of inverted repeat regions (26,090 bp) (). The base composition of the genome is 31.06% A, 31.64% T, 19.01% C, and 18.29% G, with a GC content of 37.30%. Annotation of the C. atrothea chloroplast genome identifies a total of 133 genes, including 88 protein-coding genes, 8 rRNA genes, and 37 tRNA genes (Supplemental Figures 1-2). Results also find that the C. atrothea chloroplast genome harbors a total of 72 simple sequence repeats (SSR); of them, mono-nucleotide repeats are the most abundant SSR type (53; 73.61%), followed by tetra-nucleotides (12; 16.67%), di-nucleotide (4; 5.56%), hexa-nucleotides (2; 2.78%), and tri-nucleotides (1; 1.39%) (Supplemental Table 1). Further check their polymorphism showed that 7 of them (9.72%) are polymorphic among 67 Camellia species, providing valuable genetic markers to assist tea breeding programs in the future (Supplemental Table 2).
Figure 2. The complete chloroplast genome map of C. atrothea. Arrangement of 133 genes represented in the map, including 88 protein coding genes, 37 tRNA genes, and 8 rRNA genes. The GC% along the chloroplast is represented by the inner circle.
To further examine the phylogenetic relationship between C. atrothea and other Camellia plants, we collected a total of 13 chloroplast genomes from Camellia species, and then aligned them with C. atrothea using MAFFT. Phylogenetic tree among them was then constructed using RAxML with Actinidia chinensis as outgroup and GTR + G + I as the best suitable model. Result shows that C. atrothea is close to C. leptophylla (). Both of them are clustered into a single clade comprising species from section Thea of genus Camellia, consistent with earlier findings (Wu et al. Citation2022).
Figure 3. Maximum likelihood phylogenetic tree of C. atrothea and other 14 close plant species constructed using their complete chloroplast genome sequences. The bootstrap support value for each node is shown on the branch. The accession number of chloroplast genome of each plant species is shown in the brackets.
Discussion and conclusion
We reported the 157,099 bp whole chloroplast genome sequence of C. atrothea, which encodes a total of 133 genes and comprises a large and small single-copy region as well as a pair of inverted repeats region. Read mapping shows that all genomic regions of C. atrothea chloroplast genome is well covered by sequencing reads, suggesting a high accuracy of the genome assembly (Supplemental Figure 3). Similar to many studies, the overall gene order, GC content, and gene direction of C. atrothea cp genome are comparable to those published chloroplast genomes of Camellia species such as C. sinensis (Huang et al. Citation2014). We also fully identified the polymorphic SSR in C. atrothea cp genome, and investigated the phylogenetic relationships between C. atrothea and other Camellia plants. The overall obtained results and findings will not only provide valuable genetic markers for tea breeding programs, but also offer vital phylogenetic framework for future population genetic studies and conservations in C. atrothea. In the future, we will further sequence the whole nuclear genome of C. atrothea and investigate the evolution patterns of tea quality related secondary metabolites to identify key genes associated with tea quality and disease resistance, which will help the genetic research and breeding progress of cultivated tea plants in the future.
Author contributions
E.X involved in the conception and design. Y.W. analyzed the data and wrote the article. J.H., N.X., D.Z., and W.T. interpreted the data; all authors revised the manuscript and agreed to be accountable for all aspects of the work.
Ethical approval
This research does not require Ethical approval or specific permissions according to the recommendations of the Research Ethic Committee of Anhui Agricultural University.
Supplemental Material
Download TIFF Image (14.1 MB)Supplemental Material
Download TIFF Image (4.1 MB)Supplemental Material
Download TIFF Image (15.1 MB)Supplemental Material
Download MS Word (240.7 KB)Supplemental Material
Download MS Excel (12.5 KB)Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
The assembled chloroplast genome sequence data that support the findings of this study are freely available in GenBank of NCBI (https://www.ncbi.nlm.nih.gov/) under the accession number of OK382089. The raw sequencing data of C. atrothea has also been deposited into the NCBI Bio-Project and SRA database with the accession number of PRJNA867579 and SRR20985353, respectively. The information of the plant material was deposited into NCBI Bio-Sample database with the accession number of SAMN30215288.
Additional information
Funding
References
- Beier S, Thiel T, Münch T, Scholz U, Mascher M. 2017. MISA-web: a web server for microsatellite prediction. Bioinformatics. 33(16):2583–2585.
- Chang HT. 1981. A taxonomy of the genus Camellia. Acta Scientarum Naturalium Universitatis Sunyatseni. 1(1):1–180.
- Chen S, Li RY, Ma YX, Lei SR, Ming R, Zhang XT. 2021. The complete chloroplast genome sequence of Camellia sinensis var. sinensis cultivar Tieguanyin (Theaceae). Mitochondrial DNA B Resour. 6(2):395–396.
- Cox M, Peterson D, Biggs P. 2010. SolexaQA: at-a-glance quality assessment of Illumina second-generation sequencing data. BMC Bioinformatics. 11:485.
- Daniell H, Lin C-S, Yu M, Chang W-J. 2016. Chloroplast genomes: diversity, evolution, and applications in genetic engineering. Genome Biol. 17(1):134.
- Darriba D, Taboada G, Doallo R, Posada D. 2012. jModelTest 2: more models, new heuristics and parallel computing. Nat Methods. 9(8):772.
- Huang BS. 2016. Brief of ancient tea pant resources in Yunnan province. Kunming: yunnan Fine Arts Press. p. 8–431.
- Huang H, Shi C, Liu Y, Mao SY, Gao LZ. 2014. Thirteen Camellia chloroplast genome sequences determined by high-throughput sequencing: genome structure and phylogenetic relationships. BMC Evolution Biology. 14:151.
- Jin J-J, Yu W-B, Yang J-B, Song Y, dePamphilis CW, Yi T-S, Li D-Z. 2020. GetOrganelle: a fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 21(1):241.
- Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 30(4):772–780.
- Ming TL, Bartholomew B. 2007. Theaceae in Flora of China. Wu Z. and Raven P. Beijing: science Press. 12: p. 366–478.
- Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 30(9):1312–1313.
- Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. 2013. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol. 30(12):2725–2729.
- Tillich M, Lehwark P, Pellizzer T, Ulbricht-Jones ES, Fischer A, Bock R, Greiner S. 2017. GeSeq–versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 45(W1):W6–W11.
- Wu Q, Tong W, Zhao H, Ge R, Li R, Huang J, Li F, Wang Y, Mallano AI, Deng W, et al. 2022. Comparative transcriptomic analysis unveils the deep phylogeny and secondary metabolite evolution of 116 Camellia plants. Plant J. 111(2):406–421.
- Xia EH, Yao QY, Zhang HB, Jiang JJ, Zhang LP, Gao LZ. 2015. CandiSSR: an efficient pipeline used for identifying candidate polymorphic SSRs based on multiple assembled sequences. Front Plant Sci. 6:1171.
- Yang JB, Yang SX, Li HT, Yang J, Li DZ. 2013. Comparative chloroplast genomes of Camellia species. PLoS One. 8(8):e73053.