
Abstract
Limnophila sessiliflora Blume 1826 is a perennial amphibious herb with ornamental and water purification value that is widespread in temperate and tropical Asia. In the present study, we sequenced, assembled, and annotated the complete chloroplast (cp) genome of L. sessiliflora. It is 152,395 bp in length, with a typical quadripartite structure, comprising a pair of inverted repeat regions (IRs; 25,545 bp), a large single-copy region (LSC; 83,163 bp), and a small single-copy (SSC; 18,142 bp) region. The whole cp genome contained 135 genes, including 89 protein-coding genes (PCGs), 38 transfer RNA (tRNA) genes, and eight ribosomal RNA (rRNA) genes. The maximum-likelihood (ML) phylogenetic analysis indicated that L. sessiliflora was closely related to the genera Bacopa and Scoparia in the tribe Gratioleae of Plantaginaceae. This cp genome provides a valuable genetic resource for phylogenetic study.
1. Introduction
Limnophila is a small marshy or aquatic genus with ca. 40 species, 10 of which are distributed in China (Hong et al. Citation1998). It was formerly classified in Scrophulariaceae, but now placed in the tribe Gratioleae of the family Plantaginaceae (Albach et al. Citation2005; Rahmanzadeh et al. Citation2005). Limnophila sessiliflora Blume 1826 is a perennial amphibious herb with small purplish-blue or pink axillary flowers and heteromorphic, pinnate submerged or whorled aerial leaves. Widespread in temperate and tropical Asia, this species inhabits ponds, swamps, rice fields, and wet places along streams below 1900 m (Hong et al. Citation1998), and is valuable for water purification and as an ornamental in park waterscapes and aquariums.
2. Materials and methods
The voucher specimens of L. sessiliflora used in this study were collected from Yueshan Reservoir, Qingquan Town, Xishui County, Hubei Province, China (115.2280836°E, 30.4823612°N, 35 m, in the marsh). The features of L. sessiliflora were photographed and shown in . The collected specimens were deposited in the Chinese Academy of Sciences, Herbarium of Wuhan Botanical Garden (HIB, Dr. Guangwan Hu, [email protected]), and Herbarium of Huanggang Normal University (HGTC, former Herbarium of Biology the Department of Huanggang Teachers College, Mr. Jun Fu, [email protected]) under voucher number HJD1391 collected by Dong Hongjin et al.
Figure 1. The morphology of Limnophila sessiliflora. (A) Transverse rhizome with erected branches, the aerial leaves are simple pinnate and whorled, (B) fruit, and (C) the flowers are pine, with beard in the corolla tube. All were photographed by Hongjin Dong in Xishui, the same location as the specimens collected. A and B were taken in 19 January 2021, and C was taken in 17 June 2021.

The DNA extracting and sequencing were commissioned to the company BENAGEN (Wuhan, China). Genomic DNA was extracted from fresh and tender leaves of the voucher specimen before drying, according to a modified CTAB method (Doyle and Doyle Citation1987). The concentration and quality of DNA were then detected by agarose gel electrophoresis and spectrophotometer. Finally, the qualified libraries were subjected to high-throughput sequencing by an Illumina Hiseq 2500 Sequencing System (Illumina, Hayward, CA) with 150 bp paired-end reads.
The chloroplast (cp) genome was assembled using GetOrganelle software (Jin et al. Citation2020) with default settings following guidelines. After mapping assembled cp sequence back with raw reads data, the average cover depth reached 1064Χ (Figure S1). Then, the complete cp genome of L. sessiliflora was annotated with PGA software (Qu et al. Citation2019) and manually adjusted by Geneious ver. 10.1 (http://www.geneious.com, Matthew et al. Citation2012) and then submitted to GenBank (accession number: ON000200). Genome annotation was performed by alignment with the cp genomes of related species. CPGView (Liu et al. Citation2023) was applied to visualize the gene map, the cis- and trans-splicing genes’ schematic maps of L. sessiliflora (Liu et al. Citation2023).
Ten species representing six tribes and three subfamilies of Plantaginaceae and five species representing closely related families (Mazaceae, Orobanchaceae, Paulowniaceae, Phrymaceae, and Wightiaceae) were downloaded from the NCBI database to construct a phylogenetic tree. The sequences matrix was initially aligned using MAFFT (Kazutaka and Standley Citation2013) and then manually adjusted using BioEdit (Hall Citation1999). Taking Saxifraga stolonifera Curtis 1774 (GenBank: MN496079) as an out-group, a maximum-likelihood (ML) analysis was performed with the RAxML version 8 program (Alexandros Citation2014) using 1000 bootstrap replicates. IQ-tree was also used to construct an ML tree in fast mode (Nguyen et al. Citation2015). The complete cp genome sequences of L. sessiliflora and 10 other species from Plantaginaceae and relatives were used to construct a phylogenetic tree.
3. Results
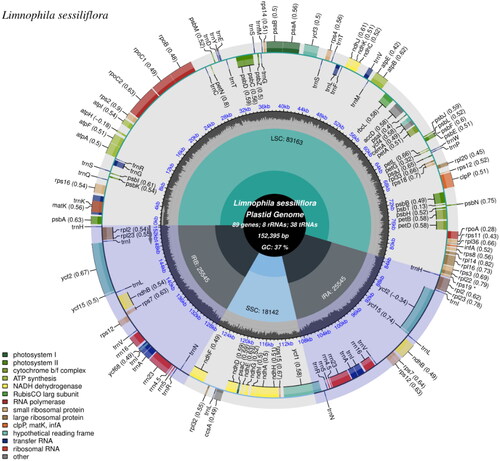
As shown in , the size of the cp genome of L. sessiliflora is 152,395 bp, including a large single-copy (LSC) region of 83,163 bp and a small single-copy (SSC) region of 18,142 bp separated by a pair of identical inverted repeat regions (IRs) of 25,545 bp each. A total of 135 genes were successfully annotated including 89 protein-coding genes (PCGs), 38 transfer RNA (tRNA) genes, and eight ribosomal RNA (rRNA) genes. The GC contents of the whole genome, IRs, LSC, and SSC regions were 37.4%, 42.9%, 35.4%, and 30.9%, respectively. The GC content of the IRs was the highest. Twenty-one genes contained one intron, while two genes had two introns. Totally, 11 cis-splicing genes (including rps16, atpF, rpoC1, ycfl, clpP1, petB, petD, rpl16, rpl2, ndhB, and ndhA, Figure S2) and one trans-splicing genes (rps12, Figure S3) were detected by CPGview (Liu et al. Citation2023).
Figure 2. The chloroplast genome map of L. sessiliflora, which was generated using CPGview (Liu et al. Citation2023). The center of the map notes the name and the length of the species, also includes the summary data of the chloroplast genome. Going outward, the first circle shows LSC, SSC, IRa, and IRb with their length. The second circle shows the GC ratio depicted as the proportion of the shaded parts of each section. The third circle displays the gene names with the colors based on their functional categories provided at the left of the circular map. Genes outside the circle are transcribed in an anticlockwise direction, and those inside are in a reverse direction.
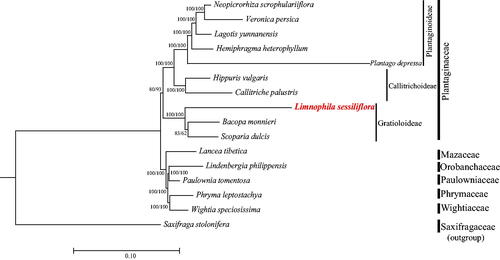
As shown in the phylogenetic tree (), L. sessiliflora belongs to the tribe Gratioleae in Plantaginaceae and is a sister group to Bacopa and Scoparia. The monophyly of the subfamilies in Plantaginaceae based on available cp genome data is highly supported, but the support value for the monophyly of Plantaginaceae was not as high as expected.
Figure 3. Maximum-likelihood phylogenetic tree including Limnophila sessiliflora based on complete chloroplast genomes. The number at each node indicates the bootstrap support value generated by RAxML/IQ-tree. The following sequences are used: Neopicrorhiza scrophulariiflora MK986819 (Zhang et al. 2019), Veronica persica KT724052 (Choi et al. 2016), Lagotis yunnanensis MN752238 (Cheng et al. 2021), Hemiphragma heterophyllum MN383192 (Wu and Zhang 2019), Plantago depressa MK144833 (Kwon et al. 2019), Hippuris vulgaris MT942637 (Liu et al. 2021), Callitriche palustris MW774642 (Yu and Dong Citation2021), Bacopa monnieri MN736955 (Liang et al. 2020), Scoparia dulcis MZ242235 (Li et al. Citation2022), Lancea tibetica MF593117 (Chi et al. 2018), Lindenbergia philippensis HG530133 (Wicke et al. 2013), Paulownia tomentosa KP718624 (Yi and Kim 2016), Phryma leptostachya MK381317 (Xia et al. 2019), Wightia speciosissima MK381318 (Xia et al. 2019), and Saxifraga stolonifera MN496079 (Dong et al. 2018). The GenBank accession numbers for each species and the citation sources for those published sequences are provided in Table S1.
4. Discussion and conclusions
The traditional recognized family Scrophulariaceae was proved to be polyphyletic, and split into some small families (Linderniaceae, Mazaceae, Paulowniaceae, and Wightiaceae), or transferred to other related families (Orobanchaceae, Phrymaceae, and Plantaginaceae), based on recent molecular evidence (Olmstead et al. Citation2001; Albach et al. Citation2005; Oxelman et al. Citation2005; Rahmanzadeh et al. Citation2005; Liu et al. Citation2020; Fowler et al. Citation2021; Mower et al. 2021). It is still phylogenetic complex between these families. The genus Limnophila together with the tribe Gratioleae, was transferred to the newly defined family Plantaginaceae, consisting of six subfamilies, 21 tribes and approximately 1760 species (Albach et al. Citation2005; Rahmanzadeh et al. Citation2005; Vito et al. Citation2022). The phylogeny of this family has attracted much attention, but only a few studies have been conducted based on the cp genome (Su et al. Citation2016; Yu and Dong Citation2021; Li et al. Citation2022). More samplings are still needed to exactly resolve the relationships between tribes and genera.
Author contributions
Conceptualization, J.Y. and H.D.; methodology and software, J.Y. and R. H.; validation and formal analysis, J.Y. and R. H.; investigation and resources, H.D.; data curation, J.Y. and S.W.; writing–original draft preparation, R. H.; writing–review and editing, J.Y., S.W., and H.D.; supervision, J.Y. and H.D.; project administration and funding acquisition, H.D. All authors read and approved the final manuscript.
Ethical approval
Limnophila sessiliflora has not been listed as national or provincial key protected wild plants in China, nor as a threatened species of the IUCN Red List. Therefore, no specific permissions or licenses were needed for the research sampling according to Regulations of the People’s Republic of China. During the sampling process, we followed the local sampling guideline for wild plants provided by Huanggang Forestry Bureau to ensure that no substantial harm to the collecting individual.
Supplemental Material
Download MS Word (24.8 KB)Supplemental Material
Download MS Word (616.4 KB)Supplemental Material
Download JPEG Image (486.1 KB){kind=link}
Supplemental Material
Download JPEG Image (1.3 MB){kind=link}
Supplemental Material
Download JPEG Image (1.2 MB){kind=link}
Acknowledgements
The authors are grateful to the editors and anonymous reviewers.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
The data that support the findings of this study are available in the NCBI GenBank database at https://www.ncbi.nlm.nih.gov under accession number ON000200. The assembled individual was linked with BioSample number SAMN26570373 and Project ID PRJNA814836. Raw sequencing reads used in this study were deposited in the GenBank Sequence Read Archive with no. SRR18313725.
Additional information
Funding
References
- Albach DC, Meudt DT, Oxelman B. 2005. Piecing together the "new" Plantaginaceae. Am J Bot. 92(2):297–315.
- Alexandros S. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 30:1312–1313.
- Doyle JJ, Doyle JL. 1987. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem Bull. 19:11–15.
- Fowler RM, Murphy DJ, McLay T, Buirchell BJ, Chinnock RJ, Bayly MJ. 2021. Molecular phylogeny of tribe Myoporeae (Scrophulariaceae) using nuclear ribosomal DNA: generic relationships and evidence for major clades. Taxon. 70(3):570–588.
- Hall TA. 1999. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp Ser. 41:95–98.
- Hong DY, Yang H, Jin CL, Holmgren NH. 1998. Scrophulariaceae. In: Wu ZY, Raven PH, editors. Flora of China. Beijing: Science Press; p. 18.
- Jin JJ, Yu WB, Yang JB, Song Y, Pamphilis C, Yi TS, Li DZ. 2020. GetOrganelle: a fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 21(1):241.
- Kazutaka K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 30(4):772–780.
- Li LX, Lin Y, Tang DF, Quan CQ, Liang Y, Qin SS, Wei F, Wei KH. 2022. The complete chloroplast genome of Scoparia dulcis (Plantaginaceae). Mitochondrial DNA B Resour. 7(1):118–119.
- Liu B, Tan YH, Liu S, Olmstead RG, Min DZ, Chen ZD, Joshee N, Vaidya BN, Chung R, Li B. 2020. Phylogenetic relationships of Cyrtandromoea and Wightia revisited: a new tribe in Phrymaceae and a new family in Lamiales. J Syst Evol. 58(1):1–17.
- Liu S, Ni Y, Li J, Zhang X, Yang H, Chen H, Liu C. 2023. CPGView: a package for visualizing detailed chloroplast genome structures. Mol Ecol Resour. 23(3):694–704.
- Matthew K, Richard M, Amy W, Steven SH, Matthew C, Shane S, Simon B, Alex C, Sidney M, Chris D. 2012. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 28:1647–1649.
- Nguyen LT, Schmidt HA, Arndt VH, Bui Quang M. 2015. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. 32(1):268–274.
- Olmstead R, Pamphilis C, Wolfe A, Young N, Elisons W, Reeves P. 2001. Disintegration of the Scrophulariaceae. Am J Bot. 88(2):348–361.
- Oxelman B, Kornhall P, Olmstead RG, Bremer B. 2005. Further disintegration of Scrophulariaceae. Taxon. 54(2):411–425.
- Qu XJ, Moore MJ, Li DZ, Yi TS. 2019. PGA: a software package for rapid, accurate, and flexible batch annotation of plastomes. Plant Methods. 15(1):1–12.
- Rahmanzadeh R, Müller K, Fischer E, Bartels D, Borsch T. 2005. The Linderniaceae and Gratiolaceae are further lineages distinct from the Scrophulariaceae (Lamiales). Plant Biol. 7(1):67–78.
- Su CK, Gi CM, Seonjoo P. 2016. The complete chloroplast genome sequences of three Veroniceae species (Plantaginaceae): comparative analysis and highly divergent regions. Front Plant Sci. 7:355.
- Vito SA, Castro SV, Mercedes S, Dalla CG, Moura MR, Olmos SA. 2022. Phylogenetics of Gratioleae (Plantaginaceae): paraphyly of Stemodia and its implications for generic circumscriptions, with insights from floral evolution. Bot J Linn Soc. 200:194–217.
- Yu JJ, Dong HJ. 2021. The complete chloroplast genome sequence of Callitriche palustris (Plantaginaceae). Mitochondrial DNA B Resour. 6(9):2777–2778.