
Abstract
Holocephali has foreseeable value to help our understanding of vertebrate genome evolution due to its phylogenetic position. In this study, we reported a complete mitochondrial genome of Hydrolagus mitsukurii, a species of holocephalans. The mitochondrial genome is 20,486 bp in length and comprised 13 PCGs, 2 rRNA, 22 tRNA, and 1 control region (D-loop), as well as a long noncoding insertion between tRNAThr and tRNAPro. A phylogenetic tree based on 13 PCGs showed that Hydrolagus mitsukurii was grouped with the members of the family Chimaeridae. Furthermore, the phylogenetic tree further supported the paraphyletic clades of Hydrolagus and Chimera.
Introduction
Hydrolagus mitsukurii (Jordan & Snyder, 1904) is a species belonging to Holocephali (chimaeras), one of the two monophyletic clades of Chondrichthyes. The species is listed as Near Threatened (NT) according to the IUCN Red List of Threatened Species 2020 (Finucci et al. Citation2020). Holocephali has foreseeable value to help our understanding of vertebrate genome evolution due to its phylogenetic position (Venkatesh et al. Citation2007; Inoue et al. Citation2010). Venkatesh et al. (Citation2007) reported the small genome (910 Mb) of an elephant shark (Callorhinchus milii), and they found that the small genome has a higher percentage of conserved sequences with the human genome compared to teleost fish genomes. A total of 15 mitogenomes of Holocephali were reported at the time of writing. The genetic information of Holocephali needs to be established to better understand the genome evolution. In this study, we sequenced the whole mitochondrial genome of H. mitsukurii to investigate the phylogenetic relationship with other species in Holocephali.
Materials and methods
A specimen of H. mitsukurii () was collected in the northern South China Sea (21.00 N 118.00 E) in August 2021 at 1300 m depth. The specimen was deposited at the Second Institute of Oceanography with voucher number B6313400002 (Dr. Xiaogu Wang [email protected]). Total DNA was extracted by Marine Animal Genomic DNA Kit (TRAN, China), and the mitochondrial genome was sequenced using the Illumina Novaseq PE250 (Shanghai Personalbioc Co., Ltd., Shanghai, China) and assembled by A5-miseq v2015052 (Coil et al. Citation2015) and SPAdes v3.9.0 (Bankevich et al. Citation2012). The collinearity of the splicing results obtained by different software was analyzed using MUMmer v3.1 (Kurtz et al. Citation2004). Based on the results of collinearity, the position relationship between contigs was determined and the gap between contigs was filled. The results were corrected using Pilon v1.18 (Walker et al. Citation2014) to gain the complete mitochondrial genome. The 5,484,646 reads were mapped in Geneious Prime (Biomatters Ltd., Auckland, New Zealand) to check the assembled mitogenome, yielding a coverage of 3,666× (Fig. S1). The complete mitogenome was annotated in MITOS Web Server (http://mitos.bioinf.uni-leipzig.de/) (Bernt et al. Citation2013). The phylogenetic analysis was performed based on 13 protein-coding genes (PCGs) from all available 15 mitogenomes of Holocephali, as well as 10 mitogenomes of sharks, and 4 mitogenomes of rays. The 13 PCGs were aligned and concatenated using Geneious Prime. The best-fit models of the evolution were calculated and maximum-likelihood phylogenetic tree was performed using IQ-Tree v1.6.12 (Kalyaanamoorthy et al. Citation2017; Minh et al. Citation2020).
Figure 1. Hydrolagus mitsukurii (Jordan & Snyder,1904), specimen B6313400002, photographed by Chen Fang (one of the authors).

Results and discussion
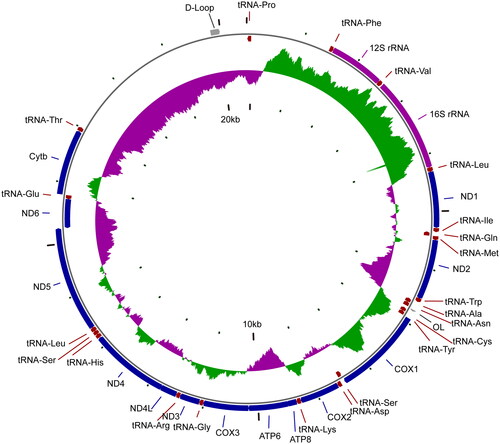
The mitochondrial genome of H. mitsukurii is 20,486 bp in length with 32.95% A, 29.43% T, 25.3% C, and 12.33% G. The genome comprised 13 PCGs, 2 ribosomal RNA genes (rRNA), 22 transfer RNA genes (tRNA) and 1 control regions (D-loop) (). All PCGs start with codon ATG and stop with TAA, except for COI and ND2. COI gene uses GTG as the start codon and terminates with the AGG codon, whereas ND2 gene terminates with AGG codon. Most genes are located on the heavy strand, with exception of 8 tRNAs and the ND6 gene which are located on the light strand. In addition, a long noncoding insertion (2843 bp) appears between tRNAThr and tRNAPro (Inoue et al. Citation2010; Gomes-dos-Santos et al. Citation2020; Gomes-dos-Santos et al. Citation2021; Vilas-Arrondo et al. Citation2022).
Figure 2. Mitogenome map of Hydrolagus mitsukurii (Jordan & Snyder, 1904).
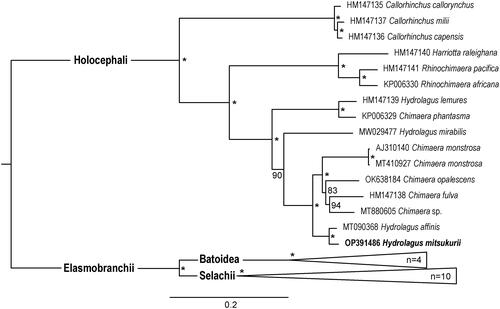
As shown in the phylogenetic tree, H. mitsukurii falls within the cluster of Holocephali and clusters with H. affinis supported by a bootstrap resampling value of 100% (). The recent studies revealed the paraphyletic status of the genus Hydrolagus and Chimera (Gomes-dos-Santos et al. Citation2020; Gomes-dos-Santos et al. Citation2021; Vilas-Arrondo et al. Citation2022). Hydrolagus and Chimera were also resolved as paraphyletic by the mitochondrial genome tree in the present study, which indicated the affinity of the two genera and the necessity of a comprehensive phylogenetic review. There is also a need to increase the genomic data to understand the phylogenetic relationship of Holocephali. The topology of the mitogenomic tree also supported the notion of a basal divergence between sharks and rays (). The mitochondrial genome of H. mitsukurii has foreseeable value in the research of evolution within Holocephali.
Figure 3. Maximum-likelihood phylogenetic tree based on the 13 PCGs showing the phylogenetic relationships of Hydrolagus mitsukurii and related taxa in Holocephali. The * above the branches indicates that bootstrap support values are above 99%. Bar 0.2 substitutions per nucleotide position.
Ethics statement
Dr. Xiaogu Wang has obtained the permission of the Second Institute of Oceanography, Ministry of Natural Resources to collect samples and experimental research since the National Key Research and Development Program. And the study complied with the ethics of the Second Institute of Oceanography, Ministry of Natural Resources.
Author contributions
Xiaogu Wang conceived and designed the experiments, Chen Fang analyzed the data and wrote the initial draft. Chunsheng Wang conceived and designed the experiments and final approval of the version to be published. All authors approved the final version of the manuscript.
Supplemental Material
Download MS Word (43 KB)Acknowledgments
We are grateful to Ms. Yuru Han for the advice in constructing the tree and to the members of R/V Yanping 2 for their help during the investigations.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
The GenBank accession number for the complete mitochondrial genome sequence of Hydrolagus mitsukurii (B6313400002) is OP391486. The BioProject and Bio-Sample and SRA numbers are PRJNA878643, SAMN30740136, and SRR21496349 respectively.
Additional information
Funding
References
- Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, et al. 2012. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 19(5):455–477. doi: 10.1089/cmb.2012.0021.
- Bernt M, Donath A, Jühling F, Externbrink F, Florentz C, Fritzsch G, Pütz J, Middendorf M, Stadler PF. 2013. MITOS: improved de novo metazoan mitochondrial genome annotation. Mol Phylogenet Evol. 69(2):313–319. doi: 10.1016/j.ympev.2012.08.023.
- Coil D, Jospin G, Darling AE. 2015. A5-miseq: an updated pipeline to assemble microbial genomes from Illumina MiSeq data. Bioinformatics. 31(4):587–589. doi: 10.1093/bioinformatics/btu661.
- Finucci B, Tanaka S, Semba Y, Kyne PM. 2020. Hydrolagus mitsukurii. The IUCN Red List of Threatened Species. 2020 e.T60194A124450319; [accessed 2023 February 28]. doi: 10.2305/IUCN.UK.2020-2.RLTS.T60194A124450319.en.
- Gomes-dos-Santos A, Arrondo NV, Machado AM, Veríssimo A, Pérez M, Román E, Castro LFC, Froufe E. 2020. The complete mitochondrial genome of the deep-water cartilaginous fish Hydrolagus affinis (de Brito Capello, 1868) (Holocephali: chimaeridae). Mitochondrial DNA. Part B. Resources. 5(2):1810–1812. doi: 10.1080/23802359.2020.1749154.
- Gomes-dos-Santos A, Vilas-Arrondo N, Machado AM, Veríssimo A, Pérez M, Baldó F, Castro LFC, Froufe E. 2021. Shedding light on the Chimaeridae taxonomy: the complete mitochondrial genome of the cartilaginous fish Hydrolagus mirabilis (Collett, 1904) (Holocephali: chimaeridae). Mitochondrial DNA B Resour. 6(2):420–422. doi: 10.1080/23802359.2020.1870887.
- Inoue JG, Miya M, Lam K, Tay BH, Danks JA, Bell J, Walker TI, Venkatesh B. 2010. Evolutionary origin and phylogeny of the modern Holocephalans (Chondrichthyes: chimaeriformes): a mitogenomic perspective. Mol Biol Evol. 27(11):2576–2586. doi: 10.1093/molbev/msq147.
- Kalyaanamoorthy S, Minh BQ, Wong TKF, von Haeseler A, Jermiin LS. 2017. ModelFinder: fast model selection for accurate phylogenetic estimates. Nat Methods. 14(6):587–589. doi: 10.1038/nmeth.4285.
- Kurtz S, Phillippy A, Delcher AL, Smoot M, Shumway M, Antonescu C, Salzberg SL. 2004. Versatile and open software for comparing large genomes. Genome Biol. 5(2):R12. doi: 10.1186/gb-2004-5-2-r12.
- Minh BQ, Schmidt HA, Chernomor O, Schrempf D, Woodhams MD, von Haeseler A, Lanfear R. 2020. IQ-TREE 2: new models and efficient methods for phylogenetic inference in the genomic era. Mol Biol Evol. 37(5):1530–1534. doi: 10.1093/molbev/msaa015.
- Venkatesh B, Kirkness EF, Loh Y, Halpern AL, Lee AP, Johnson J, Dandona N, Viswanathan LD, Tay A, Venter JC, et al. 2007. Survey sequencing and comparative analysis of the elephant shark (Callorhinchus milii) genome. PLoS Biol. 5(4):e101. doi: 10.1371/journal.pbio.0050101.
- Vilas-Arrondo N, Gomes-dos-Santos A, Pérez M, Baldó F, Veríssimo A, Catarino D, Machado AM, Román-Marcote E, Bañón R, Froufe E, et al. 2022. A mitochondrial genome assembly of the opal chimaera, Chimaera opalescens Luchetti, Iglésias et Sellos 2011, using PacBio HiFi long reads. Mitochondrial DNA B Resour. 7(3):434–437. doi: 10.1080/23802359.2022.2044403.
- Walker BJ, Abeel T, Shea T, Priest M, Abouelliel A, Sakthikumar S, Cuomo CA, Zeng Q, Wortman J, Young SK, et al. 2014. Pilon: An integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One. 9(11):e112963. doi: 10.1371/journal.pone.0112963.