
Abstract
Porella grandiloba Lindb. is a liverwort species of Porellaceae, primarily distributed in East Asia. Here, we determined the complete chloroplast (cp) genome sequence of P. grandiloba. The complete cp genome was 121,433 bp in length with a typical quadripartite structure consisting of a large single-copy region (83,039 bp), a small single-copy region (19,586 bp), and two copies of inverted repeat regions (9,404 bp, each). Genome annotation predicted 131 genes, including 84 protein-coding, 36 tRNA, and eight rRNA genes. The maximum likelihood tree indicated that P. grandiloba was sister to P. perrottetiana, which species formed a clade with Radula japonica (Radulaceae).
Introduction
Porella L. (Porellaceae) is a cosmopolitan liverwort genus comprising 120 species (Söderström et al. Citation2016; Choi et al. Citation2021). Porella species are economically important because they produce terpenoids and aromatic compounds with anticancer, antimicrobial, and antifungal activities (Asakawa Citation1998). The identification of species and analysis of phylogenetic relationships within the Porella genus are difficult owing to the high degree of variability among morphological characteristics. Porella grandiloba Lindb., 1872 is primarily distributed in East Asia including China, Japan, Republic of Korea, Taiwan, and Russian Far East (Piippo Citation1990; Bakalin and Klimova Citation2019). Natural population of P. grandiloba grows on shaded rocks and trunk base in broad-leaved forests. P. grandiloba can be distinguished from allied species by its rounded leaf dorsal lobe apices and not the decurrent ventral leaf lobes (Bakalin and Klimova Citation2019). Despite the morphological distinctiveness of species, information on chloroplast (cp) genome of P. grandiloba, which is useful for identifying species and assessing phylogenetic relationships, has not been obtained. In this study, we report the complete cp genome sequence of P. grandiloba to provide genomic information for future phylogenetic studies on related species.
Materials and methods
Leaf material of P. grandiloba was collected from Ulleungdo, Republic of Korea (37°29′15.1″N, 130°52′38.3″E). The voucher herbarium specimen was deposited at Honam National Institute of Biological Resources (HNIBR [http://en.hnibr.re.kr/]; contact person, Yongsung Kim, [email protected]) under the voucher number HNIBRMS2. The morphological images were captured using stereomicroscope Leica S9i (Leica Microsystems, Germany) and Leica DM750 optical microscope (Leica Microsystems, Germany) (). Genomic DNA was extracted using a DNeasy plant mini kit (Qiagen, Germany), according to the manufacturer’s protocol. DNA library for sequencing was constructed using a TruSeq Nano DNA library prep kit (Illumina, USA) and sequenced using a Illumina HiSeq platform (Illumina, USA). Adaptor sequences and low-quality reads were removed using Trimmomatic v0.36 (Bolger et al. Citation2014).
Figure 1. The morphological images of Porella grandiloba. (A) Dorsal view of a specimen plant and (B) median cells of a leaf were captured using stereomicroscope (Leica S9i) and light microscope (Leica DM750), respectively.

The high-quality reads were assembled for the complete cp genome using GetOrgnelle (Jin et al. Citation2020). Annotation was predicted using GeSeq (Tillich et al. Citation2017) and manually corrected using Geneious Prime 2022.2.2 (http://www.geneious.com) comparing to chloroplast genome sequences of closely relative species. To ensure the accuracy of the assembly, we examined the depth of coverage by mapping the reads that were employed in de novo assembly process (Supplementary Figure S1). The circular map of the chloroplast genome was generated using OGDRAW ver.1.3.1 (Greiner et al. Citation2019). The structures of intron-containing genes were visualized using CPGview (Liu et al. Citation2023). The complete chloroplast sequence of P. grandiloba was deposited in GenBank of the National Center for Biotechnology Information (NCBI) with accession OP476656.
To determine the phylogenetic position, we included 11 species from five families (Lejeuneaceae [seven species], Jubulaceae [one], Frullaniaceae [one], Radulaceae [one], and Porellaceae [one]) of Porellales in addition to P. grandiloba. We also included Apopellia endiviifolia (Pelliaceae; Pelliales) and Marchantia polymorpha (Marchantiaceae; Marchantiales) as outgroups. The data of cp genomes were downloaded from GenBank. The concatenated alignment sequences of 82 protein-coding genes were extracted from complete cp genomes. The sequences were aligned using MAFFT (Katoh et al. Citation2019). Phylogenetic tree was constructed based on the maximum likelihood (ML) method and 1000 bootstrap replicates using IQ-TREE (Nguyen et al. Citation2015).
Results and discussion
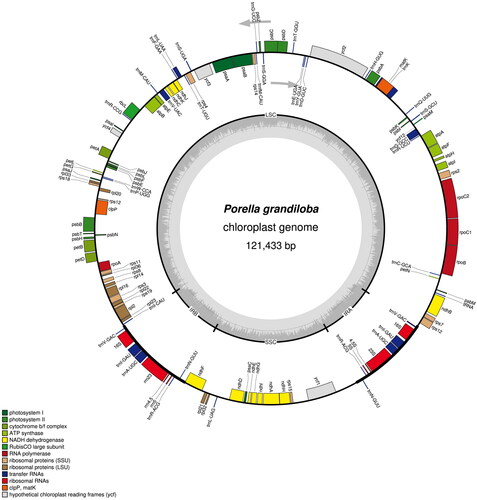
The complete cp genome of P. grandiloba was 121,433 bp in length (). The cp genome was contained typical quadripartite structure, containing a large single-copy region of 83,039 bp, a small single-copy region of 19,586 bp, and a pair of inverted repeat regions of 9,404 bp each. The overall GC content was 33.7%. The cp genome annotation predicted a total of 128 genes including 84 protein-coding, 36 tRNA, and eight rRNA genes. There are 10 pretein-coding genes (ndhB, rpoC1, atpF, ycf3, clpP, petB, petD, rpl16, rpl2, and ndhA) containing intron(s) and one trans-splicing genes rps12 (Supplementary Figure S2). In total, nine genes replicate in the IR region, repeating inversely with each other, including five tRNA genes (trnV-GAC, trnI-GAU, trnA-UGC, trnR-ACG, and trnN-GUU) and four rRNA genes (rrn16S, trn23S, trn4.5S, and trn5S).
Figure 2. The circular map of the chloroplast genome of Porella grandiloba generated using OGDRAW (Greiner et al. Citation2019). the large single-copy (LSC) and small single-copy (SSC) are separated by inverted repeat (IRs; IRA and IRB). the genes inside the circular map are transcribed clockwise and outside are transcribed counterclockwise. The genes with related functions are shown in the same color. Built-in gray histogram represents the GC content of the genome and the gray line in the Middle represents the threshold of 50%.
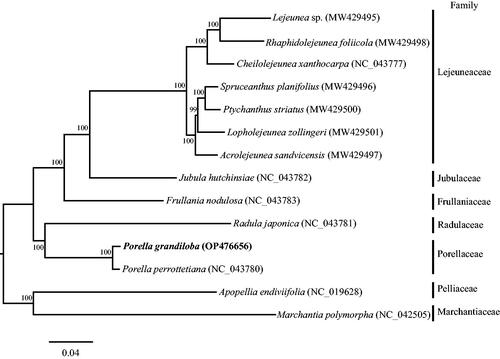
A phylogenetic tree was constructed using IQTREE, based on the principle of maximum likelihood estimate, with cp protein-coding DNA sequences from 12 representative species of order Porellales (). The ML phylogenetic tree indicated that P. grandiloba was sister to P. perrottetiana (Mont.) Trevis., which species formed a clade with Radula japonica (Radulaceae). As the sequence data for Porellaceae cp genomes is limited, more research is needed to understand the phylogenetic relationship within this family. In order to gain a deeper understanding of the evolutionary history of P. grandiloba, it is necessary to obtain more complete chloroplast sequences from other Porella species.
Figure 3. Maximum likelihood tree based on 82 protein-coding gene sequences of 14 complete chloroplast genomes. Apopellia endiviifolia and Marchantia polymorpha were used as outgroups. The GenBank accession number has been provide within parentheses after the species name. Numbers above the branches indicate bootstrap supporting values based on 1000 replicates. The bar represents 0.04 nucleotide substitution per site.
This study examined the complete cp genome of P. grandiloba for the first time using Illumina sequencing. The cp genome data of P. grandiloba is expected to contribute valuable information for molecular indentification and elucidation of phylogenetic relationships among the different species of Porellaceae.
Ethics statement
This article does not involve studies with human participants or animals. The species in this paper is not endangered, protected, or personally owned.
Authors’ contribution
SAL, CK, and KJL conceptualized and designed the study; SAL, S-JP, and HS performed sample collection, laboratory work, and bioinformatic analyses; SAL and S-JP prepared the draft; CK and KJL revised the draft. All authors approved the final version of the manuscript and agreed to be accountable for all aspects of the work.
Supplemental Material
Download MS Word (103 KB)Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
The chloroplast genome sequence data that support the findings of this study are publicly available in GenBank of NCBI (http://www.ncbi.nlm.nih.gov) under the accession number OP476656. The associated BioProject, BioSample, and SRA numbers are PRJNA886845, SAMN31143293, and SRR21796986, respectively.
Additional information
Funding
References
- Asakawa Y. 1998. Biologically active compoounds from bryophytes. J Hattori Bot Lab. 84:91–104.
- Bakalin VA, Klimova KG. 2019. Porellaceae (Hepaticae) in the Russian Far East. Bot Pac. 8(1):105–131. doi:10.17581/bp.2019.08110.
- Bolger AM, Lohse M, Usadel B. 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 30(15):2114–2120. doi:10.1093/bioinformatics/btu170.
- Choi SS, Bakalin V, Park SJ. 2021. Integrating continental mainland and islands in temperate East Asia: liverworts and hornworts of the Korean Peninsula. PhytoKeys. 176:131–226. doi:10.3897/phytokeys.176.56874.
- Greiner S, Lehwark P, Bock R. 2019. OrganellarGenomeDRAW (OGDRAW) version 1.3.1: expanded toolkit for the graphical visualization of organellar genomes. Nucleic Acids Res. 47(W1):W59–W64. doi:10.1093/nar/gkz238.
- Jin JJ, Yu WB, Yang JB, Song Y, dePamphilis CW, Yi TS, Li DZ. 2020. GetOrganelle: a fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 21(1):241. doi:10.1186/s13059-020-02154-5.
- Katoh K, Rozewicki J, Yamada KD. 2019. MAFFT online service: multiple sequence alignment, interactive sequence choice and visualization. Brief Bioinform. 20(4):1160–1166. doi:10.1093/bib/bbx108.
- Liu S, Ni Y, Li J, Zhang X, Yang H, Chen H, Liu C. 2023. CPGView: a package for visualizing detailed chloroplast genome structures. Mol Ecol Resour. 0:1–11.
- Nguyen LT, Schmidt HA, von Haeseler A, Minh BQ. 2015. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. 32(1):268–274. doi:10.1093/molbev/msu300.
- Piippo S. 1990. Annotated catalogue of Chinese Hepaticae and Anthocerotae. J Hattori Bot Lab. 68:1–192.
- Söderström L, Hagborg A, von Konrat M, Bartholomew-Began S, Bell D, Briscoe L, Brown E, Cargill DC, Costa DP, Crandall-Stotler BJ, et al. 2016. World checklist of hornworts and liverworts. PK. 59:1–828. doi:10.3897/phytokeys.59.6261.
- Tillich M, Lehwark P, Pellizzer T, Ulbricht-Jones ES, Fischer A, Bock R, Greiner S. 2017. GeSeq – versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 45(W1):W6–W11. doi:10.1093/nar/gkx391.