
Abstract
The first complete mitochondrial genome of the dart sac-bearing camaenid Laeocathaica Möllendorff, Citation1899 was sequenced and analyzed in this study. The whole mitogenome of Laeocathaica amdoana Möllendorff, Citation1899 was 14,660 bp in length and its nucleotide composition showed high AT-content of 67.45%. It had 37 genes, including 13 protein-coding genes, two ribosomal RNA genes, and 22 transfer RNA genes. The phylogeny yielded by both Bayesian inference and maximum-likelihood method suggested that Laeocathaica was closely related to the other dart sac-bearing camaenids with known complete mitochondrial genome. These genetic data are expected to provide fundamental resources for further genetic studies on the camaenids.
1. Introduction
Laeocathaica amdoana Möllendorff, Citation1899 belongs to the genus Laeocathaica Möllendorff, Citation1899, a group of dart sac-bearing camaenids (Möllendorff Citation1899). L. amdoana is characterized by a typical Laeocathaica sinistral shell and anatomically, two tiny proximal accessory sacs and each has an opening leading to the dart sac chamber. This species is endemic to NW Sichuan and S Gansu, China (Wu et al. Citation2023).
At present, the public nucleotide database has no information pertaining to the mitogenome of this genus. For the first time, we sequenced, annotated, and analyzed the complete mitochondrial genome of L. amdoana, which might provide new data for the reconstruction of the camaenid phylogeny.
2. Materials
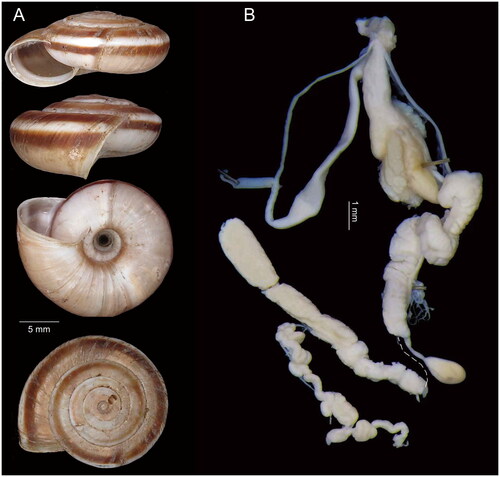
The specimen (voucher no. HBUMM08456, ) was collected from Shichuanba, Wenxian County, Gansu Province (33.17534°N, 105.019362°E) and deposited in Hebei University (contact Min Wu: [email protected]). The reference images were prepared using a Canon camera. Species identification was based on shell morphology () and genital anatomy according to the diagnostic characteristics proposed by Wu et al. (Citation2023). The elongated part between atrium and dart sac apparatus and two tiny proximal accessory sacs () make this species clearly different from those species with similar shells in the genus Laeocathaica, i.e. L. distinguenda Möllendorff, Citation1899 (in which the part between atrium and dart sac is not elongated) and L. tropidorhaphe Möllendorff, Citation1899 (in which the proximal accessory sac is absent) and is obviously L. amdoana.
Figure 1. Species reference images of Laeocathaica amdoana Möllendorff, Citation1899 (voucher no. HBUMM08456).
3. Methods
3.1. DNA extraction, sequencing, and assembly
We sequenced the mitogenome of L. amdoana using the next-generation sequencing (NGS) techniques. Genomic DNA was extracted using CTAB method. Raw data were generated using Illumina Novaseq6000 platform. The lengths of reads and inserted sequence are 2 × 150 bp and 400 bp, respectively. Software fastp v0.36 (Chen et al. Citation2018) was applied to filter raw data. After quality control (QC), the clean reads were assembled via SPAdes v3.15 (Bankevich et al. Citation2012). Then, we used blastn (Altschul et al. Citation1997) to compare scaffolds with those existing sequences in the NT database (NCBI) to investigate sequence similarity. GapFiller v1.11 (Boetzer and Pirovano Citation2012) was used to supplement gaps. The sequences were checked and corrected using PrInSeS-G (Massouras et al. Citation2010) before the complete circular mitogenome sequence was obtained.
3.2. Annotation of mitogenome
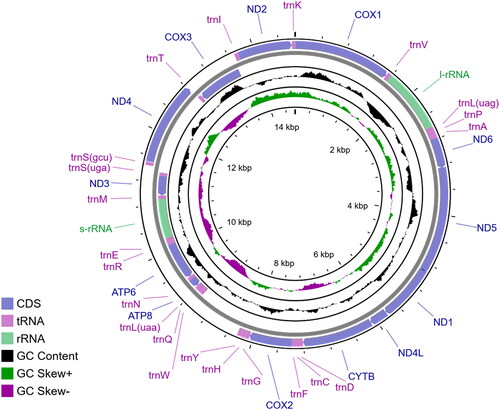
The limits of protein-coding genes (PCGs) were adjusted with aids of genewise (Birney et al. Citation2004) and tblastn (Altschul et al. Citation1997) against the mitogenomic sequences of other assumed related species. Ribosomal RNA genes (rRNAs) were predicted using cmsearch (Nawrocki and Eddy Citation2013) based on the CM models constructed by mitos (Bernt et al. Citation2013). Transfer RNA genes (tRNAs) were identified with MiTFi (Li et al. Citation2013). After predicting the gene positions by softwares, we manually checked and adjusted the boundaries of every gene according to the criteria mentioned by both Fourdrilis et al. (Citation2018) and Ghiselli et al. (Citation2021). We used a circos plot () to display the annotation results.
Figure 2. The mitochondrial genome of Laeocathaica amdoana Möllendorff, Citation1899, HBUMM08456 (OP866270). Arrows indicate the directions of transcription.
3.3. Phylogenetic analysis
Complete mitochondrial genomes of two species of Helicidae and 11 camaenids including that sequenced by this work, all known so far, were obtained from NCBI for reconstructing the phylogeny of Camaenidae. We use helicids Helix pomatia Linnaeus, 1758 and Cylindrus obtusus Draparnaud, 1805 as the outgroups, considering that phylogenetically Helicidae and Bradybaenidae (sensu Wade et al. Citation2001) (=Bradybaeninae) are almost the nearest relatives based on 5.8S gene, the complete internal transcribed spacer 2 (ITS2) region, and approximately 840 nucleotides of the large subunit (28S) gene (Wade et al. Citation2001, Citation2007). In addition, the mitochondrial genome of camaenid Euhadra herklotsi was sequenced in 1997, and was submitted to GenBank in nine separate parts (see ), instead of a complete circular genome, so we synthesized these nine records to obtain 37 genes (for details of the complete mitogenome that we organized, see supplementary material 1).
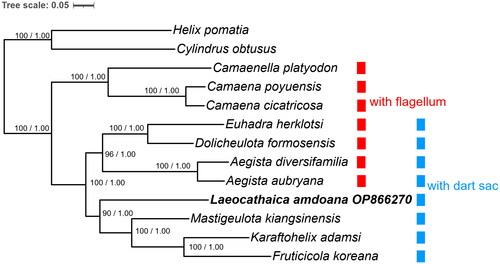
Figure 3. Bayesian phylogram based on the 1st and 2nd codons of eight unsaturated PCGs and two rRNA genes, with Helicidae as outgroup. Numbers near nodes indicate Bayesian posterior probabilities (BPP) and maximum-likelihood bootstrap (BP) values, given as BP/BPP. The mitogenome of L. amdoana determined in this study is highlighted in bold. Scale bar is for substitutions per site. The following sequences were used: Aegista diversifamilia KR002567 and Dolicheulota formosensis KR338956 (Huang et al. Citation2016), Camaena poyuensis KT001074 (Lin et al. Citation2016), Fruticicola koreana KU237291 (unpublished), Karaftohelix adamsi KY230382 (unpublished), Camaenella platyodon MH362759 (unpublished), Cylindrus obtusus JN107636 (Groenenberg et al. Citation2012), Mastigeulota kiangsinensis KM083123 (Deng et al. Citation2016), Camaena cicatricosa KM365408 (Wang et al. Citation2014), Aegista aubryana KT192071 (Yang et al. Citation2016), Helix pomatia MK488031 (Groenenberg and Duijm Citation2019), and Euhadra herklotsi Z71693–Z71701 (Yamazaki et al. Citation1997).
The extraction and alignment of PCGs and rRNA genes were performed with PhyloSuite v1.2.3 (Xiang et al. Citation2023). Ambiguously aligned fragments of 13 PCG alignments were removed in batches using Gblocks 0.91b (Talavera and Castresana Citation2007) with default parameter settings, and gap sites of rRNA genes were removed with trimAl v1.2rev57 (Capella-Gutiérrez et al. Citation2009) using ‘-automated1’ command. DAMBE v7.3.32 (Xia Citation2018) was employed to make the saturation test for every gene of PCGs and rRNAs. Unsaturated sequences were concatenated in the same order for subsequent analyses. Best-fit partition model (Edge-linked, ) was selected under BIC criterion using ModelFinder (Kalyaanamoorthy et al. Citation2017). Bayesian inference phylogenies were inferred using MrBayes 3.2.7a (Ronquist et al. Citation2012) under partition model (two parallel runs, 530,000 generations), in which the initial 25% of the sampled data were discarded as burn-in. Maximum-likelihood phylogenies were inferred using IQ-TREE (Nguyen et al. Citation2015) under Edge-linked partition model for 5000 ultrafast (Minh et al. Citation2013) bootstraps.
Table 1. The best-fitting model of each partition selected for MrBayes and IQ-tree.
4. Results
4.1. Mitogenomic characterization
The average sequence coverage was 39.34×. The complete mitochondrial genome of L. amdoana was circular and 14,660 bp in length. The nucleotide composition was 28.68%, 38.77%, 18.18%, and 14.37% for A, T, G, and C, respectively. This mitogenome contained 37 genes, including 13 PCGs, two rRNA genes, and 22 tRNA genes. Among them, 23 genes were transcribed along the forward direction and the rest along the reverse direction. A few genes were overlapped with their neighboring genes. The length of gene overlaps in the whole mitochondrial genome was 40 bp. All PCGs used ATK as the start codon except COX2 and NAD3 starting with TTG, and CYTB starting with GTG. In addition, all PCGs use TAR as stop codon, except COX2 ending with an abbreviated stop codon T, and ATP6 and ATP8 ending with TA. The length of tRNA genes ranged from 53 to 63 bp. Intergenic regions that contained 952 bp in total ranged between 1 and 816 bp, accounting for 6.50% of the whole mitogenome. The longest intergenic region, also an AT-rich region, was located between trnWuca and trnYgua with a 67.89% AT content.
4.2. Phylogenetic analysis
The saturation tests indicated that ATP8, NAD2, NAD3, NAD4L, and NAD6 are saturated genes. The 1st and 2nd codons of the rest PCGs and two rRNA genes were therefore concatenated for phylogenetic analyses. The phylogenies reconstructed by Bayesian and ML methods were topologically identical (). It indicated that L. amdoana was sister to the clade (Mastigeulota kiangsinensis, (Karaftohelix adamsi, Fruticicola koreana)). The clade of dart sac-bearing camaenids was divided into two groups based on the status of flagellum. The camaenids without dart sac apparatus are basal in the clade including all the examined camaenids.
5. Discussion and conclusions
The proposed phylogeny () coincides with that by the phylogeny based on the 13 PCGs on the positions of Mastigeulota Pilsbry, 1895, Euhadra Pilsbry, 1890 and Camaena Albers, 1850 (Wang et al. Citation2014). The phylogenetic result () generally agrees with the phylograms based on the sequence data of 16S and COX1 (Chen et al. Citation2021), where Camaeninae that has no dart sac apparatus and the dart sac-bearing Bradybaeninae, with exception of Pseudostegodera Wu et Chen, Citation2021, Stegodera Martens, 1876, and Nesiohelix Kuroda et Emura 1943, appeared as sister taxa. The topology where the monophyly Dolicheulota Pilsbry, 1901 + Euhadra (1.0 BPP and 100% BP) and Aegista Albers, 1850 are sister taxa (), is consistent with the 16S + COX1-based phylogeny proposed by Jirapatrasilp et al. (Citation2022).
In Laeocathaica, the change of the gene order, in comparison to those in the other camaenid taxa, was observed (for details, see supplementary material 2). The gene order of Laeocathaica agrees with that of Mastigeulota on the same branch, and Euhadra and Dolicheulota on the sister branch. The present work confirms that the evolution of flagellum and dart sac apparatus might have played significant roles in the course of formation of camaenid diversity ().
By this work, we reported the mitogenome of L. amdoana for the first time, which is congruent with the typical mitochondrial genome structure in metazoan (Saccone et al. Citation1999). The systematic position of Laeocathaica within Camaenidae was first inferred based on complete mitogenomes, and these results would provide fundamental resources for further studies on the evolution of this genus and the phylogenetics of Camaenidae.
Author contributions
WS prepared figures, analyzed data, uploaded data to NCBI, and drafted the paper. MW designed this work, identified the species, drafted and approved the final draft. They both agree to be accountable for this work.
Ethical approval
This research did not involve ethical research because snails are exempt from ethical approval or permission.
Supplemental Material
Download (13 KB)Supplemental Material
Download (29.1 KB)Supplemental Material
Download TIFF Image (1.6 MB)Acknowledgements
We are grateful to Mr. Qiming Li, who collected the specimen for this work. Sequencing services were provided by Personal Biotechnology Co., Ltd., Shanghai. Gene assembly and annotation services were provided by Sangon Biotech.
Disclosure statement
No potential conflict of interest was reported by the authors, and the authors alone are responsible for the content and writing of the paper.
Data availability statement
The GenBank ID of this genome sequence is OP866270. The BioProject, BioSample, and SRA numbers of metadata are PRJNA912872, SAMN32260481, and SRR22762582, respectively. The CM models for predicting rRNA genes can be downloaded on GitLab at https://gitlab.com/Bernt/MITOS/tree/master/data/modelle-v1.
Additional information
Funding
References
- Altschul SF, Madden TL, Schaffer AA, Zhang JH, Zhang Z, Miller W, Lipman DJ. 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25(17):3389–3402. doi: 10.1093/nar/25.17.3389.
- Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, et al. 2012. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 19(5):455–477. doi: 10.1089/cmb.2012.0021.
- Bernt M, Donath A, Jühling F, Externbrink F, Florentz C, Fritzsch G, Pütz J, Middendorf M, Stadler PF. 2013. MITOS: improved de novo metazoan mitochondrial genome annotation. Mol Phylogenet Evol. 69(2):313–319. doi: 10.1016/j.ympev.2012.08.023.
- Birney E, Clamp M, Durbin R. 2004. GeneWise and genomewise. Genome Res. 14(5):988–995. doi: 10.1101/gr.1865504.
- Boetzer M, Pirovano W. 2012. Toward almost closed genomes with GapFiller. Genome Biol. 13(6):R56. doi: 10.1186/gb-2012-13-6-r56.
- Capella-Gutiérrez S, Silla-Martínez JM, Gabaldón T. 2009. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics. 25(15):1972–1973. doi: 10.1093/bioinformatics/btp348.
- Chen S, Zhou Y, Chen Y, Gu J. 2018. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics. 34(17):i884–i890. doi: 10.1093/bioinformatics/bty560.
- Chen Z-Y, Lyu Z-T, Wu M. 2021. Systematic revision of Stegodera Martens, 1876 (Gastropoda, Stylommatophora, Camaenidae), with description of a new genus. Zookeys. 1059:1–21. doi: 10.3897/zookeys.1059.68385.
- Deng P-J, Wang W-M, Huang X-C, Wu X-P, Xie G-L, Ouyang S. 2016. The complete mitochondrial genome of Chinese land snail Mastigeulota kiangsinensis (Gastropoda: Pulmonata: Bradybaenidae). Mitochondrial DNA A DNA Mapp Seq Anal. 27(2):1441–1442. doi: 10.3109/19401736.2014.953083.
- Fourdrilis S, de Frias Martins AM, Backeljau T. 2018. Relation between mitochondrial DNA hyperdiversity, mutation rate and mitochondrial genome evolution in Melarhaphe neritoides (Gastropoda: Littorinidae) and other Caenogastropoda. Sci Rep. 8(1):17964. doi: 10.1038/s41598-018-36428-7.
- Ghiselli F, Gomes-dos-Santos A, Adema CM, Lopes-Lima M, Sharbrough J, Boore JL. 2021. Molluscan mitochondrial genomes break the rules. Philos Trans R Soc Lond B Biol Sci. 376(1825):20200159. doi: 10.1098/rstb.2020.0159.
- Groenenberg DSJ, Duijm E. 2019. The complete mitogenome of the Roman snail Helix pomatia Linnaeus 1758 (Stylommatophora: Helicidae). Mitochondrial DNA Part B. 4(1):1494–1495. doi: 10.1080/23802359.2019.1601512.
- Groenenberg DSJ, Pirovano W, Gittenberger E, Schilthuizen M. 2012. The complete mitogenome of Cylindrus obtusus (Helicidae, Ariantinae) using Illumina next generation sequencing. BMC Genomics. 13(1):114. doi: 10.1186/1471-2164-13-114.
- Huang C-W, Lin S-M, Wu W-L. 2016. Mitochondrial genome sequences of landsnails Aegista diversifamilia and Dolicheulota formosensis (Gastropoda: Pulmonata: Stylommatophora). Mitochondrial DNA A DNA Mapp Seq Anal. 27(4):2793–2795. doi: 10.3109/19401736.2015.1053070.
- Jirapatrasilp P, Huang C-W, Hwang C-C, Sutcharit C, Lee C-T. 2022. Convergent evolution of Amphidromus-like colourful arboreal snails and phylogenetic relationship of East Asian camaenids, with description of a new Aegistohadra species (Helicoidei: Camaenidae: Bradybaeninae). Invertebr Syst. 36(3):244–290. doi: 10.1071/IS21015.
- Kalyaanamoorthy S, Minh BQ, Wong TKF, von Haeseler A, Jermiin LS. 2017. ModelFinder: fast model selection for accurate phylogenetic estimates. Nat Methods. 14(6):587–589. doi: 10.1038/nmeth.4285.
- Li X-J, Yang J, Wang J-H, Ren Q-L, Li X, Huang Y. 2013. Methods and software tools for mitochondrial genome assembly and annotation. Chin J Appl Entomol. 50:298–304.
- Lin J-H, Zhou W, Ding H-L, Wang P, Ai H-M. 2016. The mitochondrial genome of the land snail Cernuella virgata (Da Costa, 1778): the first complete sequence in the family Hygromiidae (Pulmonata, Stylommatophora). ZooKeys. 589:55–69. doi: 10.3897/zookeys.589.7637.
- Massouras A, Hens K, Gubelmann C, Uplekar S, Decouttere F, Rougemont J, Cole ST, Deplancke B. 2010. Primer-initiated sequence synthesis to detect and assemble structural variants. Nat Methods. 7(7):485–486. doi: 10.1038/nmeth.f.308.
- Minh BQ, Nguyen MAT, von Haeseler A. 2013. Ultrafast approximation for phylogenetic bootstrap. Mol Biol Evol. 30(5):1188–1195. doi: 10.1093/molbev/mst024.
- Möllendorff OF. 1899. Binnen-Mollusken aus Westchina und Centralasien. I. Annuaire du Musée Zoologique de L’Académie Impériale Des St.-Pétersburg. 4:46–144.
- Nawrocki EP, Eddy SR. 2013. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics. 29(22):2933–2935. doi: 10.1093/bioinformatics/btt509.
- Nguyen L-T, Schmidt HA, von Haeseler A, Minh BQ. 2015. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. 32(1):268–274. doi: 10.1093/molbev/msu300.
- Ronquist F, Teslenko M, van der Mark P, Ayres DL, Darling A, Hohna S, Larget B, Liu L, Suchard MA, Huelsenbeck JP. 2012. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol. 61(3):539–542. doi: 10.1093/sysbio/sys029.
- Saccone C, De Giorgi C, Gissi C, Pesole G, Reyes A. 1999. Evolutionary genomics in Metazoa: the mitochondrial DNA as a model system. Gene. 238(1):195–209. doi: 10.1016/s0378-1119(99)00270-x.
- Talavera G, Castresana J. 2007. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst Biol. 56(4):564–577. doi: 10.1080/10635150701472164.
- Wade CM, Hudelot C, Davison A, Naggs F, Mordan PB. 2007. Molecular phylogeny of the helicoid land snails (Pulmonata: Stylommatophora: Helicoidea), with special emphasis on the Camaenidae. J. Molluscan Stud. 73(4):411–415. doi: 10.1093/mollus/eym030.
- Wade CM, Mordan PB, Clarke B. 2001. A phylogeny of the land snails (Gastropoda: Pulmonata). Proc Biol Sci. 268(1465):413–422. doi: 10.1098/rspb.2000.1372.
- Wang P, Yang H, Zhou W, Hwang C, Zhang W, Qian Z. 2014. The mitochondrial genome of the land snail Camaena cicatricosa (Müller, 1774) (Stylommatophora, Camaenidae): the first complete sequence in the family Camaenidae. ZooKeys. 451:33–48. doi: 10.3897/zookeys.451.8537.
- Wu M, Shen W, Chen Z-G. 2023. Land snail diversity in central China: revision of Laeocathaica Möllendorff, 1899 (Gastropoda, Camaenidae), with descriptions of seven new species. Zookeys. 1154:49–147. doi: 10.3897/zookeys.1154.86237.
- Xia X. 2018. DAMBE7: new and improved tools for data analysis in molecular biology and evolution. Mol Biol Evol. 35(6):1550–1552. doi: 10.1093/molbev/msy073.
- Xiang C-Y, Gao F, Jakovlić I, Lei H-P, Hu Y, Zhang H, Zou H, Wang G-T, Zhang D. 2023. Using PhyloSuite for molecular phylogeny and tree-based analyses. iMeta. 2(1):e87. doi: 10.1002/imt2.87.
- Yamazaki N, Ueshima R, Terrett JA, Yokobori S, Kaifu M, Segawa R, Kobayashi T, Numachi K, Ueda T, Nishikawa K, et al. 1997. Evolution of pulmonate gastropod mitochondrial genomes: comparisons of gene organizations of Euhadra, Cepaea and Albinaria and implications of unusual tRNA secondary structures. Genetics. 145(3):749–758. doi: 10.1093/genetics/145.3.749.
- Yang X, Xie G-L, Wu X-P, Ouyang S. 2016. The complete mitochondrial genome of Chinese land snail Aegista aubryana (Gastropoda: Pulmonata: Bradybaenidae). Mitochondrial DNA A DNA Mapp Seq Anal. 27(5):3538–3539. doi: 10.3109/19401736.2015.1074207.