
Abstract
Water flea Ceriodaphnia dubia has been widely used for risk assessments of chemicals and environmental contamination. In this study, the complete mitochondrial genome (mitogenome) of this species NIES strain was determined using short-read high throughput and long-read sequencing technologies. The mitogenome of C. dubia was 15,170 bp in length and consisted of 13 protein-coding genes (PCGs), 2 ribosomal RNAs (rRNAs), and 22 transfer RNAs (tRNAs). The gene order was identical to the pattern conserved across crustaceans. The complete mitogenome of the NIES strain will serve as genetical reference in ecological risk assessments in Japan, as well as resources for future phylogenetical studies using cladocerans.
Introduction
The cladocerans are pelagic water fleas and have been widely used in ecotoxicological studies and chemical risk assessments. In particular, Daphnia magna Straus 1820, D. pulex Leydig 1860, Ceriodaphnia dubia Richard 1894, and Moina macrocopa Straus 1820 are representative and have been cultured in many laboratories worldwide. National Institute for Environmental Studies (NIES), Japan has established the culture of these cladoceran species (i.e. NIES strains) and provided the NIES strains to laboratories throughout Japan (https://www.nies.go.jp/kenkyu/yusyo/suisei/index.html) for risk assessments of chemicals and environmental contamination (Mano et al. Citation2010; Hayasaka et al. Citation2012; Watanabe et al. Citation2016). Despite its ecotoxicological importance, C. dubia () does not have a published complete mitochondrial genome for any strains. In this study, we report the complete mitogenome sequences of C. dubia, NIES strain. The complete mitogenome of the NIES strain is beneficial as a genetic resource for future ecotoxicological research as well as to investigate intrageneric phylogenetic relationships.
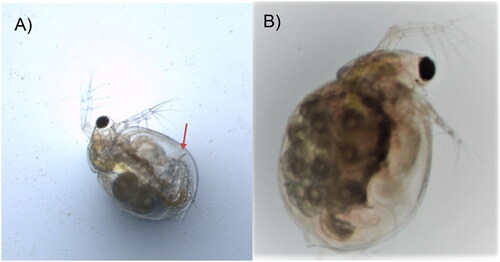
Figure 1. Photos of Ceriodaphnia dubia. This species can be distinguished from other related species based on dorsolateral depression between head and rest of body, (A) postadominal claw (arrow), and (B) lack of rostrum and tail spine. The photos were taken by the authors.
Materials
The specimens of C. dubia were obtained from laboratory culture that was maintained at the NIES for more than 30 years. The laboratory culture originated from 15 individuals that were transported from the United States Environmental Protection Agency in 1994. Notably, since their initial transportation in 1994, there has been no further introduction from external sources, indicating the uniformity of this lineage.
Methods
DNA was extracted from whole body using DNeasy Blood & Tissue Kit (Qiagen, Hilden, Germany) and sequenced on an Illumina MiSeq with a paired-end library. The extracted DNA was deposited in the Ecotoxicity Research Section at the NIES, Japan (NIES-202202-CLA1; Kyoshiro Hiki, [email protected]). The sequence data were processed and assembled using CLC Genomics Workbench version 22.0 following the method described in our previous study (Hiki et al. Citation2021). Low coverage region between 12S rRNA and trnQ was amplified using primers (Forward: 5′-GCTGGCACGATTTTGGTCAAT-3′, Reverse: 5′-GGCATGAGCCCACTAGCTTA-3′) and additionally sequenced using Oxford Nanopore Technologies MinION with SQK-RAD004 kit. We filtered out reads with low-quality scores by NanoFilt version 2.8.0 (De Coster et al. Citation2018), with parameters of–headcrop 10 and–quality 10. To fill the gap in the draft assembly, we mapped the Nanopore long-reads to the draft assembly and used Pilon version 1.24 (Walker et al. Citation2014) to integrate these reads with the Illumina short-reads. The accuracy of the assembly was confirmed by visualizing the read coverage depth with integrative genomics viewer (IGV) (Thorvaldsdóttir et al. Citation2013). The mitogenome was annotated by MITOS 2 webserver (Bernt et al. Citation2013) and by manual comparison with orthologous genes of other daphnid species. The circular mitogenome was visualized using Proksee (Grant et al. Citation2023).
Phylogenetic analysis was performed based on 13 protein-coding gene (PCG) sequences in the mitogenome and those of other selected cladocerans within the infraorder Anomopoda. Each amino acid sequence was aligned separately using MAFFT version 7.490 with the L-INS-i option (Katoh and Standley Citation2013) and then the alignments were filtered using trimAl version 1.4.1 (Capella-Gutiérrez et al. Citation2009) with the heuristic method. Maximum likelihood-based phylogenetic trees were inferred using IQ-TREE version 2.1.2 (Nguyen et al. Citation2015) with the ‘–p’ option to allow partition-specific evolution rates and visualized by the ggtree R package version 3.4.4 (Yu et al. Citation2017). The best-fit model for each PCG was determined by ModelFinder (Kalyaanamoorthy et al. Citation2017) implemented in IQ-TREE, based on Akaike information criteria.
Results
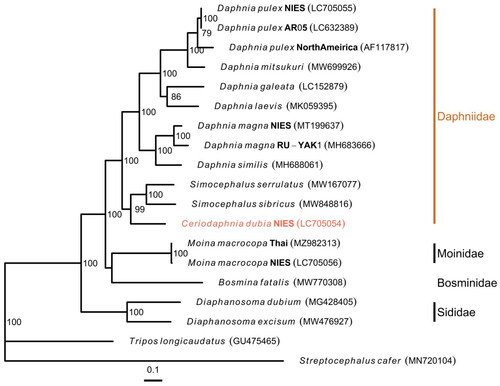
The complete circular mitogenome of C. dubia NIES strain (INSDC accession numbers: LC705054.2) was 15,170 bp in length (). The mitogenome contained typical gene components, including 13 PCGs, two ribosomal RNAs (rRNAs), and 22 transfer RNAs (tRNAs). The order of the genes was completely identical to that reported for the mitogenome of D. pulex (Crease Citation1999). The ratio of A + T nucleotides was 69.5%. All 13 PCGs initiated with the standard start codons (i.e. ATN and TTG). Whereas most of all PCGs terminated with complete stop codons (i.e. TAA and TAG), several PCGs had termination codons consisting of only T: cox1, cox2, and nad5. Putative control region located between 12S rRNA and trnQ, and contained two different inverted repeats (47 and 41 bp with GC content of 23.4 and 26.8%, respectively). Although the Illumina reads had low depth of coverage in the putative control region (>20×), likely due to its low GC content, the Nanopore reads provided sufficient depth (>21,000×) (Figure S1), indicating a highly confident assembly. The maximum likelihood-based phylogenetic analysis showed that C. dubia was grouped with Simocephalus genus (Gu et al. Citation2021) with a high bootstrap value and that the Daphniidae family formed the monophyletic group within the infraorder Anomopoda ().
Figure 2. Circular map of the mitochondrial genome of C. dubia. Genes outside the circle are encoded on the heavy strand and genes inside the circle are encoded on the light stand. The inner black bars indicate the GC content, and the Middle line represents 50%. Visualization was performed using Proksee (Grant et al. Citation2023).
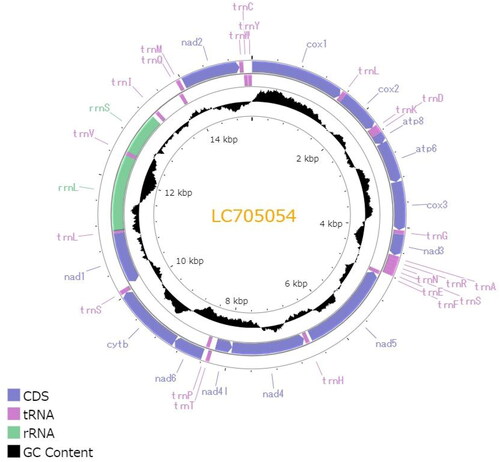
Figure 3. Maximum likelihood Tree based on 13 PCGs in mitochondrial genomes of cladocerans. Red species represent the mitogenomes obtained in this study. S. cafer and T. longicaudatus were used as the outgroup. Non-parametric bootstrap values (based on 3000 times resampling) are shown at nodes. The scale bar indicates the number of amino acid substitutions per site. The following sequences were used to infer the tree: Daphnia pulex LC705055.1, D. pulex LC632389.1 (Ohtsuki et al. Citation2022), D. pulex AF117817.1 (Crease Citation1999), D. mitsukuri MW699926.1, D. galeata LC152879.1 (Tokishita et al. Citation2017), D. laevis MK059395.1 (Martins Ribeiro et al. Citation2019), D. magna MT199637.1 (Lee et al. Citation2020), D. magna MH683666.1 (Fields et al. Citation2018), D. similus MH688061.1 (Fields et al. Citation2018), Simocephalus serrulatus MW167077.1, S. sibricus MW848816.1 (Gu et al. Citation2021), C. dubia LC705054.2 (this study), Moina macrocopa MZ982313.1 (Nam et al. Citation2022), M. macrocopa LC705056.1, bosmina fatalis MW770308.1 (Wei et al. Citation2021), Diaphanosoma dubium MG428405.1 (Liu et al. Citation2017), Diaphanosoma excisum MW476927.1 (Pan et al. Citation2021), tripos longicaudatus GU475465.1 (Ryu and Hwang Citation2010), and streptocephalus cafer MN720104.1 (Tladi et al. Citation2020).
Discussion and conclusion
This study presents the first report of the complete mitogenome of C. dubia NIES strain. The obtained mitogenome exhibited typical gene components and gene order. Phylogenetic analysis confirmed that C. dubia belonged to Daphniidae family. These results, along with the sequence data, establish a valuable genetic resource for the NIES strain of this species, which will contribute to ecotoxicological research in Japan.
Ethical approval
This study does not involve ethical issues. The species used in this study is not endangered organisms, and the sampling site is not located in any protected area.
Author contributions
K.H., H.W., H.Y., and T.Y. conceived and designed the project. K.O. performed sample collection and DNA extraction. K.O, H.W., and H.Y. contributed to the culture maintenance. N.N. and K.H. performed sequencing experiments. K.H. analyzed the data and wrote the draft manuscript. All authors reviewed and approved the final manuscript.
Supplemental Material
Download MS Word (110.4 KB)Acknowledgments
The authors are grateful to Prof. Norihisa Tatarazako (Ehime University) for the establishment of C. dubia NIES strain. The authors are grateful to anonymous reviewers and an editor for their useful comments that greatly improved the quality of the manuscript.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
Data that support the findings of this study are openly available at INSDC with the accession number LC705054.2, and at Sequence Read Archives (SRA) with the accession number PRJDB13090 (BioProject), SAMD00444315 (C. dubia) (BioSample), DRX338201 and DRX338203 (Experiment), and DRR352290 and DRR352292 (Run). The sequences at two different SRA accession numbers were derived from the same DNA sample but from different sequencing runs.
Additional information
Funding
References
- Bernt M, Donath A, Jühling F, Externbrink F, Florentz C, Fritzsch G, Pütz J, Middendorf M, Stadler PF. 2013. MITOS: improved de novo metazoan mitochondrial genome annotation. Mol Phylogenet Evol. 69(2):313–319. doi: 10.1016/j.ympev.2012.08.023.
- Capella-Gutiérrez S, Silla-Martínez JM, Gabaldón T. 2009. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics. 25(15):1972–1973. doi: 10.1093/bioinformatics/btp348.
- Crease TJ. 1999. The complete sequence of the mitochondrial genome of Daphnia pulex (Cladocera: Crustacea). Gene. 233(1–2):89–99. doi: 10.1016/s0378-1119(99)00151-1.
- De Coster W, D’Hert S, Schultz DT, Cruts M, Van Broeckhoven C. 2018. NanoPack: visualizing and processing long-read sequencing data. Bioinformatics. 34(15):2666–2669. doi: 10.1093/bioinformatics/bty149.
- Fields PD, Obbard DJ, McTaggart SJ, Galimov Y, Little TJ, Ebert D. 2018. Mitogenome phylogeographic analysis of a planktonic crustacean. Mol Phylogenet Evol. 129:138–148. doi: 10.1016/j.ympev.2018.06.028.
- Grant JR, Enns E, Marinier E, Mandal A, Herman EK, Chen C, Graham M, Van Domselaar G, Stothard P. 2023. Proksee: in-depth characterization and visualization of bacterial genomes. Nucleic Acids Res. 51(W1):W484–W492. doi: 10.1093/nar/gkad326.
- Gu YL, Liu P, Han BP. 2021. Complete mitochondrial genome of freshwater flea Simocephalus sibiricus Sars, 1899 (Crustacea: Cladocera: Anomopoda). Mitochondrial DNA B Resour. 6(11):3100–3102. doi: 10.1080/23802359.2021.1981787.
- Hayasaka D, Korenaga T, Suzuki K, Sánchez-Bayo F, Goka K. 2012. Differences in susceptibility of five cladoceran species to two systemic insecticides, imidacloprid and fipronil. Ecotoxicology. 21(2):421–427. doi: 10.1007/s10646-011-0802-2.
- Hiki K, Oka K, Nakajima N, Yamamoto H, Yamagishi T, Sugaya Y. 2021. The complete mitochondrial genome of the non-biting midge Chironomus yoshimatsui (Diptera: Chironomidae). Mitochondrial DNA B Resour. 6(10):2995–2996. doi: 10.1080/23802359.2021.1975511.
- Kalyaanamoorthy S, Minh BQ, Wong TKF, von Haeseler A, Jermiin LS. 2017. ModelFinder: fast model selection for accurate phylogenetic estimates. Nat Methods. 14(6):587–589. doi: 10.1038/nmeth.4285.
- Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 30(4):772–780. doi: 10.1093/molbev/mst010.
- Lee JS, Kim DH, Choi BS, Kato Y, Watanabe H, Lee JS. 2020. Complete mitochondrial genome of the freshwater water flea Daphnia magna NIES strain (Cladocera, Daphniidae): rearrangement of two ribosomal RNA genes. Mitochondrial DNA Part B. 5(2):1822–1823. doi: 10.1080/23802359.2020.1750995.
- Liu P, Xu S, Huang Q, Dumont HJ, Lin Q, Han BP. 2017. The mitochondrial genome of Diaphanosoma dubium with comparison with Daphnia magna. Mitochondrial DNA B Resour. 2(2):926–927. doi: 10.1080/23802359.2017.1413295.
- Mano H, Sakamoto M, Tanaka Y. 2010. A comparative study of insecticide toxicity among seven cladoceran species. Ecotoxicology. 19(8):1620–1625. doi: 10.1007/s10646-010-0547-3.
- Martins Ribeiro M, Facchin S, Pereira AH, Kalapothakis E, Xu S, Han BP, Dumont HJ, Cecília Rietzler A. 2019. Mitogenome of Daphnia laevis (Cladocera, Daphniidae) from Brazil. Mitochondrial DNA Part B Resour. 4(1):194–196. doi: 10.1080/23802359.2018.1545547.
- Nam SE, Kim J, Rhee JS. 2022. First complete mitochondrial genome from family Moinidae, Moina macrocopa (Straus, 1820) (Cladocera; Moinidae). Mitochondrial DNA B Resour. 7(6):980–982. doi: 10.1080/23802359.2022.2080024.
- Nguyen LT, Schmidt HA, von Haeseler A, Minh BQ. 2015. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. 32(1):268–274. doi: 10.1093/molbev/msu300.
- Ohtsuki H, Norimatsu H, Makino T, Urabe J. 2022. Invasions of an obligate asexual daphnid species support the nearly neutral theory. Sci Rep. 12(1):7305. doi: 10.1038/s41598-022-11218-4.
- Pan J, Liu P, Pajk F, Dumont HJ, Han BP. 2021. The mitochondrial genome of Diaphanosoma excisum Sars, 1885 (Crustacea: Branchiopoda: Cladocera) from Hainan Island, China. Mitochondrial DNA B Resour. 6(3):1279–1280. doi: 10.1080/23802359.2021.1907252.
- Ryu JS, Hwang UW. 2010. Complete mitochondrial genome of the longtail tadpole shrimp Triops longicaudatus (Crustacea, Branchiopoda, Notostraca). Mitochondrial DNA. 21(5):170–172. doi: 10.3109/19401736.2010.503809.
- Thorvaldsdóttir H, Robinson JT, Mesirov JP. 2013. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform. 14(2):178–192. doi: 10.1093/bib/bbs017.
- Tladi M, Dalu T, Rogers DC, Nyamukondiwa C, Parbhu SP, Teske PR, Emami-Khoyi A, Wasserman RJ. 2020. The complete mitogenome of the fairy shrimp Streptocephalus cafer (Lovén, 1847) (Crustacea: Branchiopoda: Anostraca) from an ephemeral pond in Botswana, southern Africa. Mitochondrial DNA B Resour. 5(1):623–625. doi: 10.1080/23802359.2019.1711222.
- Tokishita S, Ichi Shibuya H, Kobayashi T, Sakamoto M, Ha JY, Yokobori S i, Yamagata H, Hanazato T. 2017. Diversification of mitochondrial genome of Daphnia galeata (Cladocera, Crustacea): comparison with phylogenetic consideration of the complete sequences of clones isolated from five lakes in Japan. Gene. 611:38–46. doi: 10.1016/j.gene.2017.02.019.
- Walker BJ, Abeel T, Shea T, Priest M, Abouelliel A, Sakthikumar S, Cuomo CA, Zeng Q, Wortman J, Young SK, et al. 2014. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One. 9(11):e112963. doi: 10.1371/journal.pone.0112963.
- Watanabe H, Tamura I, Abe R, Takanobu H, Nakamura A, Suzuki T, Hirose A, Nishimura T, Tatarazako N. 2016. Chronic toxicity of an environmentally relevant mixture of pharmaceuticals to three aquatic organisms (alga, daphnid, and fish). Environ Toxicol Chem. 35(4):996–1006. doi: 10.1002/etc.3285.
- Wei W, Zhang K, Shi Q. 2021. Complete mitochondrial genome of Bosmina fatalis (Cladocera: Bosminidae) and its phylogenetic analysis. Mitochondrial DNA B Resour. 6(9):2567–2568. doi: 10.1080/23802359.2021.1959442.
- Yu G, Smith DK, Zhu H, Guan Y, Lam TTY. 2017. Ggtree: an R package for visualization and annotation of phylogenetic trees with their covariates and other associated data. Methods Ecol Evol. 8(1):28–36. doi: 10.1111/2041-210X.12628.